|
|
|


来氟米特活性代谢物特立氟胺即A771726控制高血糖 治疗糖尿病的机制扬州大学兽医学院研究进展?潜力?
关注者
5
被浏览
5,149
登录后你可以
不限量看优质回答
私信答主深度交流
精彩内容一键收藏

先看文献原文
基于S6K1靶点的抗2型糖尿病和抗肥胖的药物作用机理研究--《扬州大学》2018年博士论文 (cnki.com.cn) [1] 中国知网 (cnki.net) [2] (wanfangdata.com.cn) [3] 百度学术 [4] [5] [6]
【摘要】: 糖尿病是人类最重要的一种慢性代谢性疾病。生活水平的提高和生活方式的改变导致的营养过剩和运动缺乏是诱发2型糖尿病和肥胖的最重要因素。微血管和大血管循环障碍带来的糖尿病综合症是糖尿病发病和死亡的主要原因。病理生理学研究发现糖尿病的发生主要是由于胰岛素分泌不足和受体敏感性下降,即胰岛素抵抗。在正常情况下,胰岛素结合至胰岛素受体能够激活下游的磷脂酰肌醇-3激酶(PI-3K)信号通路。该通路中的一个关键性激酶是AKT,激活AKT能够促进葡萄糖转运蛋白4(GLUT4)从细胞浆转移至细胞膜上,从而增加葡萄糖吸收。另外,AKT能促进进入细胞内的葡萄糖合成为糖原;AKT同时还能抑制肝细胞内的糖异生从而减少葡萄糖释放。在胰岛素抵抗的情况下,胰岛素刺激其受体的敏感性下降,导致AKT的磷酸化水平增加不明显,AKT活化能力不足,调控血糖水平能力下降从而导致血糖升高,即糖尿病。S6K1(p70 S6激酶)是PI-3K通路中的一个重要的丝氨酸/苏氨酸激酶,该酶在营养过剩的情况下持续性过度激活,显著降低胰岛素受体的敏感性,诱发2型糖尿病和肥胖。S6K1下游的一个重要底物是胰岛素受体底物(IRS-1)。S6K1磷酸化IRS-1的S1101位点,降低IRS-1的稳定性,抑制IRS-1与磷脂酰肌醇-3激酶(PI-3K)的p85亚单位的结合。抑制S6K1活性会反馈性激活PI-3K信号通路,增加葡萄糖吸收。S6K1在糖尿病的发病机理中发挥重要的作用,S6K1抑制剂可增加胰岛素受体的敏感性从而增加葡萄糖吸收并降低血糖水平。S6K1在肥胖的发病机理中也起着非常重要的作用。S6K1基因缺陷型的小鼠能够抵抗高脂肪饲料诱导的肥胖和2型糖尿病。一方面主要是由于S6K]参与脂肪干细胞的分化;另一方面,近年来研究发现在肿瘤细胞和神经元细胞中,S6K1抑制剂抑制S6K1活性后,导致AMPK激活进而诱导自噬。在脂肪细胞中,含脂滴的自噬小体与溶酶体融合后将对甘油三酯进行降解。鉴于此,我们推测S6K1抑制剂可能会抑制脂肪细胞的分化并通过诱导自噬促进脂滴的降解。近来研究发现抗类风湿性关节炎(RA)药物来氟米特(leflunomide)及其活性代谢产物A77 1726能抑制S6K1活性。由于来氟米特是一个人工合成的化合物,本研究拟从植物抽提的化合物中遴选出一个S6K1抑制剂并研究其是否具有调控血糖的效果。我们对发表的文献进行搜索,发现生姜提取物姜烯酮A(GinA)是一个S6K1抑制剂。本研究以2型糖尿病小鼠模型、肌管细胞和脂肪细胞为研究对象,使用这两个S6K1抑制剂,通过蛋白免疫印迹、免疫共沉淀、免疫荧光、葡萄糖耐受试验(GTT)和胰岛素耐受试验(ITT)等技术手段,揭示S6K1抑制剂对2型糖尿病和肥胖中关键信号蛋白和功能的影响,以期阐明S6K1抑制剂治疗2型糖尿病和肥胖的临床前药效及其分子机制,为抗糖尿病和抗肥胖药物的研发提供理论依据和新思路。具体研究分为以下三个部分:1.来氟米特的降血糖作用及其机理研究为研究来氟米特对2型糖尿病小鼠的降血糖作用及其分子机制,我们在体外试验中以3T3-LI脂肪细胞、C2C12和L6肌管细胞为细胞模型,用来氟米特的活性代谢产物A77 1726处理细胞,运用蛋白免疫印迹检测胰岛素信号通路和PI-3K通路相关蛋白的磷酸化水平;运用免疫共沉淀检测PI-3激酶的p85亚基与IRS-1的结合;运用免疫荧光检测GLUT4的移位情况,同时运用同位素示踪检测葡萄糖的吸收情况。在体内试验中我们建立了高脂肪饮食(HFD)诱导的2型糖尿病小鼠模型,并且应用了 ob/ob小鼠,在来氟米特灌胃3d后,检测小鼠血糖水平并分别进行GTT和ITT测定,同时检测ob/ob小鼠体内AKT的磷酸化水平。结果表明,A77 1726能增加AKTS473/T308和S6KIT389的磷酸化水平,并降低S6S235/236和IRS-IS1101的磷酸化水平。A77 1726能增加胰岛素受体的酪氨酸磷酸化水平以及PI-3激酶p85亚基与IRS-1的结合。在L6细胞中,A77 1726能增强胰岛素刺激的GLUT4转移至细胞膜上。在L6肌管细胞和3T3-L1脂肪细胞中,A77 1726能增强胰岛素刺激的葡萄糖吸收。ob/ob和HFD饲喂的小鼠口服灌胃来氟米特后,小鼠血糖恢复到正常水平并能克服GTT和ITT中的胰岛素抵抗,且对正常饮食饲喂的小鼠没有影响。来氟米特能增加ob/ob小鼠脂肪和肌肉中AKTS473/T308的磷酸化水平,但对正常小鼠无影响。本研究表明来氟米特通过抑制S6K1活性来致敏胰岛素受体,并且来氟米特对于治疗同时患有RA和糖尿病的患者具有潜在的意义。2.Gin A通过抑制S6K1致敏胰岛素受体并增加葡萄糖吸收为阐明Gin A对葡萄糖吸收的作用及其分子机制,我们以3T3-L1脂肪细胞和L6肌管细胞为细胞模型,用GinA处理细胞,通过蛋白免疫印迹检测信号通路相关蛋白的磷酸化水平,用免疫共沉淀检测PI-3激酶的p85亚基与IRS-1的结合;运用免疫荧光检测GLUT4的移位情况,同时用2-NBDG化学发光法检测葡萄糖的吸收情况。蛋白免疫印迹结果显示,Gin A能增加AKTS473和S6KIT389的磷酸化水平并降低S6S235/236和IRS-1S1101的磷酸化水平,表明Gin A能反馈性激活PI-3激酶。蛋白免疫印迹和免疫共沉淀结果显示,Gin A能增加胰岛素受体的酪氨酸磷酸化以及PI3K的p85亚基与IRS-1的结合。免疫荧光试验结果表明,Gin A能增强胰岛素诱导的GLUT4移位至细胞膜上。2-NBDG荧光测定结果显示Gin A能增强3T3-L1脂肪细胞和L6肌管细胞中胰岛素刺激的葡萄糖吸收。本研究表明,Gin A通过抑制S6K1致敏胰岛素受体并增加体外葡萄糖吸收。Gin A对糖尿病有潜在的治疗作用。3.来氟米特以自噬-溶酶体途径促进脂质代谢为揭示来氟米特对脂质代谢的作用及其作用机理,我们在体外以3T3-L1脂肪细胞为细胞模型,用来氟米特的活性代谢产物A77 1726处理细胞,运用蛋白免疫印迹检测自噬相关蛋白的表达水平以及AMPK信号通路蛋白的磷酸化水平;运用免疫荧光检测自噬小体与溶酶体、自噬小体与脂滴的共定位情况;同时运用脂滴油红染色检测脂滴降解情况。蛋白免疫印迹结果显示,A77 1726能显著增加自噬标志物LC3 II的表达,并以时间和剂量依赖方式增加AMPKT172和ULKS555的磷酸化水平。通过Bafilomycin阻断自噬小体和溶酶体结合、GFP-RFP-LC3双荧光标记以及自噬小体和溶酶体的共定位试验,表明A77 1726能激活AMPK并诱导完全自噬。脂滴油红染色和自噬小体与脂滴共定位试验结果表明,A771726介导的自噬通过溶酶体降解脂滴。本研究表明,A77 1726通过激活3T3-L1脂肪细胞AMPK并以自噬-溶酶体途径调节脂质代谢。
关键词: 来氟米特;A77 1726;GinA;高血糖;肥胖;S6K1;
- 专辑: 医药卫生科技
- 专题: 药学
- 分类号: R965
导师: 徐秀龙;
学科专业: 预防兽医学
博士电子期刊出版信息: 年期:2019年第01期网络出版时间:2018-12-16——2019-01-15
Control of hyperglycemia in male mice by leflunomide: mechanisms of action in: Journal of Endocrinology Volume 237 Issue 1 (2018) [通过来氟米特控制雄性小鼠高血糖:作用机制:内分泌学杂志第237卷第1期(2018)] (bioscientifica.com) [7] https://doi.org/10.1530/JOE-17-0536 [8] PDF下载 [9] 百度学术 [10]
| 以下谷歌翻译未经校对,仅供专业人士校正用。较多错误请以原文为准 |
|---|
来氟米特控制雄性小鼠的高血糖:作用机制 在
内分泌学杂志
Authors:
Junhong Chen
1
,
2
,
Jing Sun
1
,
2
,
Michelle E Doscas
3
,
Jin Ye
3
,
Ashley J Williamson
4
,
Yanchun Li
5
,
Yi Li
6
,
Richard A Prinz
7
, and
Xiulong Xu
1
,
2
,
3
,
8
- 1 扬州大学比较医学研究所, 江苏省扬州市
- | 2 扬州大学兽医学院, 江苏省扬州
- | 3 美国伊利诺伊州芝加哥拉什大学医学中心细胞与分子医学系
- | 4 拉什医学院,拉什大学医学中心,芝加哥,伊利诺伊州,美国
- | 5 美国伊利诺伊州芝加哥大学医学系内分泌科
- | 6 美国德克萨斯州休斯顿贝勒医学院莱斯特和苏史密斯乳房中心
- | 7 美国伊利诺伊州埃文斯顿北岸大学卫生系统外科
-
| 8 扬州大学江苏省重大动物传染病与人畜共患病防控协同创新中心,扬州
DOI: https:// doi.org/10.1530/JOE-17- 0536 卷/期: 第 237 卷:第 1 期 页面范围: 43–58
文章类型: 研究文章网上出版日期: 2018 年 4 月版权: © 2018 内分泌学会 2018
自由登入 -
下载PDF
检查更新 - 获取权限
- 摘要/摘录
- 全文
- PDF格式
- 补充材料
摘要
p70 S6 激酶 (S6K1) 是一种丝氨酸/苏氨酸激酶,可磷酸化胰岛素受体底物 1 (IRS-1) 的丝氨酸 1101 并使胰岛素受体信号转导脱敏。由于营养过剩导致的 S6K1 过度激活导致高血糖和 2 型糖尿病。我们最近的研究表明,抗类风湿性关节炎 (RA) 药物来氟米特的活性代谢物 A77 1726 是 S6K1 的抑制剂。来氟米特是否能控制高血糖和使胰岛素受体敏感尚未得到检验。在这里,我们报告 A77 1726 增加了 AKT S473/T308和 S6K1 T389磷酸化,但降低了 S6 S235/236和 IRS-1 S11013T3-L1 脂肪细胞、C2C12 和 L6 肌管中的磷酸化。A77 1726 增加胰岛素受体酪氨酸磷酸化和 PI-3 激酶的 p85 亚基与 IRS-1 的结合。A77 1726 增强了 L6 肌管和 3T3-L1 脂肪细胞中胰岛素刺激的葡萄糖摄取,并增强了胰岛素刺激的 4 型葡萄糖转运蛋白 (GLUT4) 易位至 L6 细胞的质膜。 最后,我们研究了来氟米特对ob/ob 和高脂饮食 (HFD) 诱导的糖尿病小鼠模型的抗高血糖作用。 在ob/ob 和 HFD 喂养的小鼠中,来氟米特治疗使血糖水平正常化并克服了葡萄糖和胰岛素耐受试验中的胰岛素抵抗,但对喂养正常食物 (NCD) 的小鼠没有影响。来氟米特治疗增加 AKT S473/T308 ob/ob 小鼠的脂肪和肌肉中的磷酸化,但在正常小鼠中没有。我们的研究结果表明,来氟米特通过 在体外 抑制 S6K1 活性使胰岛素受体敏感,并且来氟米特可能对治疗 RA 和糖尿病患者有用。
关键词: 来氟米特 ; p70 S6 激酶 ; 胰岛素抵抗 ; 胰岛素受体底物 ; 高血糖
介绍
2 型糖尿病是一个主要的公共卫生问题(f="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib43">Zimmet等人, 2001 年,
2014 年
)。尽管有许多抗糖尿病药物可供使用,但有些药物在长期使用后会产生无法忍受的副作用或失去治疗效果(
Nathan 2015
)。未能控制高血糖会导致糖尿病并发症,这是大多数糖尿病相关发病率和死亡率的原因(
Nathan 2015
)。由于缺乏运动和循环炎症细胞因子,患有类风湿性关节炎 (RA) 的糖尿病患者可能比没有 RA 的患者出现更严重的高血糖症(
Herlitz-Cifuentes等人2015 年
,ef="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib24">Pinto等人2017 年))。与没有 RA 的人相比,患有 RA 的人患 2 型糖尿病 ( f="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib19">Jiang et al. 2015 ) 和肥胖症 (
"https://joe.bioscientifica.com/configurable/content/journals$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib35">Versini et al. 2014
) 的风险明显更高。目前,RA 和糖尿病患者分别接受抗糖尿病和抗 RA 药物治疗。一种同时控制 RA 和高血糖的药物可以极大地使患有这两个问题的患者受益。
胰岛素与其受体的结合激活了胰岛素受体酪氨酸激酶,导致胰岛素受体自身磷酸化和细胞内蛋白质底物如胰岛素受体底物 (IRS) 的磷酸化 (
图 1B
) (
Guo 2013
,
2014
)。酪氨酸磷酸化的 IRS 与 PI-3 激酶的 p85 亚基相互作用并激活其催化性 p110 亚基(
图 1B
)。PI-3 激酶激活导致丝氨酸磷酸化和蛋白激酶 B (AKT) 的激活 (
Guo 2013
,
2014
)。AKT 激活在葡萄糖代谢中起关键作用(ref="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib9">Dann等人2007 年,
Copps & White 2012 年)
)。AKT 激活通过诱导 4 型葡萄糖转运蛋白 (GLUT4) 易位至脂肪和肌肉细胞的质膜来刺激葡萄糖摄取(
图 1
)(
Guo 2013
,
2014
)。此外,AKT还通过刺激糖原合成和抑制糖异生来调节糖代谢(
图1B
)(
Guo 2013
,
2014
)。
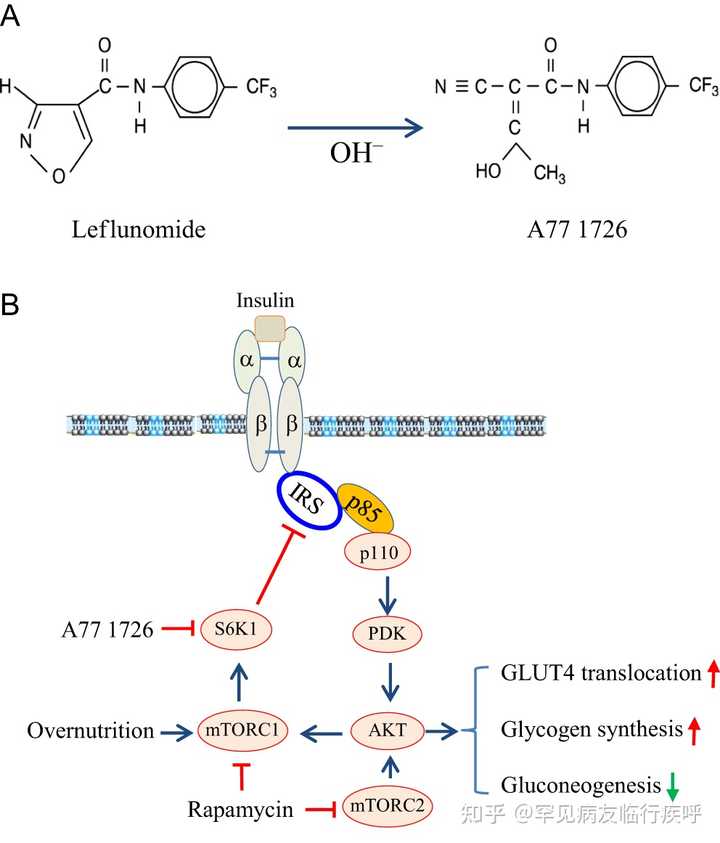

图1
来氟米特介导的抗高血糖作用机制。(A) 来氟米特和 A77 1726 的化学结构。(B) A77 1726 的作用方式。高浓度脂肪酸和氨基酸的营养过剩导致组成型 S6K1 活化,使 IRS-1 S1101磷酸化,导致 AKT 活化不良。来氟米特及其活性代谢物 A77 1726 抑制 S6K1 活性,随后导致 IRS-1 S1101的抑制。IRS-1 S1101磷酸化的抑制导致胰岛素受体敏化,这可以通过增加胰岛素受体酪氨酸磷酸化和增加 IRS-1 与 PI-3 激酶的 p85 亚基的结合来揭示。AKT S473/T308磷酸化和活化通过刺激 GLUT4 膜易位、增加糖原合成和减少糖异生而导致葡萄糖摄取增加。长期使用雷帕霉素会抑制 mTORC1 和 mTORC2,从而加剧高血糖。此图的全彩版本可在 https:// doi.org/10.1530/JOE-17- 0536 获得。
引文:内分泌学杂志 237, 1; 10.1530/JOE-17-0536
- 下载图
-
将图下载为 PowerPoint 幻灯片
雷帕霉素 (mTOR) 激酶的机制靶标是由 AKT 激活的丝氨酸/苏氨酸激酶。高浓度氨基酸和脂肪酸的营养过剩也会激活 mTOR( 图 1B )。p70 S6 激酶 (S6K1),一种 mTOR 下游的丝氨酸/苏氨酸蛋白激酶,磷酸化 IRS 并随后减弱 PI3K 通路的激活( 图 1B )( Fenton & Gout 2010 )。高胰岛素血症或营养过剩导致的本构 S6K1 激活导致胰岛素受体脱敏( Boura-Halfon & Zick 2009 , Copps & White 2012 )。S6K1 还参与调节几种能量消耗相关基因的表达,例如黑皮质素 4 受体 (MC4R) ( href=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib34">Um等。2004 , 2006 ,< a href=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib37">夏等。2012 年)。S6K1 -/-小鼠在喂食高脂肪饮食 (HFD) 时不会出现肥胖和高血糖症 (ref=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib34"> Um et al. 2004 )。这些小鼠的寿命明显长于野生型小鼠 ( "https://joe.bioscientifica.com/configurable/content/journals$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib29"> Selman et al. 2009 )。胰岛素受体信号在喂食 HFD 的 S6K1 -/-小鼠的代谢组织中高度活跃,其肝脏、肌肉和脂肪中的 AKT 磷酸化增加就证明了这一点(href=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib34"> Um等人, 2004 年))。S6K1 是在营养过剩的情况下驱动胰岛素抵抗和诱导肥胖的关键激酶 ( ef=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib9">Dann et al. 2007 )。
来氟米特是一种口服前药,被禁止用于治疗类风湿性关节炎 (RA) ( Breedveld & Dayer 2000 )。消化不良后,它会在胃肠道、血浆和肝脏中迅速且完全(>99%)转化为其活性代谢物 A77 1726( 图 1A )( Breedveld & Dayer 2000 )。一旦进入血浆,A77 1726 就会与血浆蛋白紧密结合,主要是白蛋白 ( Cannon & Kremer 2004 )。A77 1726 的半衰期很长,为 15.5 天(范围 14-18 天),在代谢成三氟甲基苯胺-草酸 (60-70%) 并排泄到尿液中后被清除 ( Breedveld & Dayer 2000 , Cannon &克雷默 2004 )。A77 1726 是来氟米特唯一的活性代谢物,完全负责其治疗活性。 A77 1726 抑制 蛋白质 酪氨酸 激酶 和 二氢 乳清 酸 脱氢酶 ( _ _ _ _ 。_ 活性 的 ) DHase - DHO _ _ . . . A77 1726 抑制DHO -DHase 活性的能力 (IC50)。大约 100 nM 的值)比其抑制蛋白酪氨酸激酶(如p56 lck、p59 fyn和 PDGF 受体)的活性(IC 50值大约为 25-50 µM)的能力强约 10-100 倍( href=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib40">Xu等人) 1995 年、 1996 年 、 1997 年 、 1999 年 , Ruckemann等人 1998 年 )。来氟米特抑制嘧啶核苷酸合成被认为是其主要作用机制( Williamson et al. 1996 , "https://joe.bioscientifica.com/configurable/content/journals$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib3">Bruneau et al. 1998 )。然而,外源性尿苷的添加使 体外 细胞培养物中的嘧啶核苷酸水平正常化,只能部分拮抗这种抗增殖作用。 在 MRL/MpJ- lpr/lpr 小鼠的淋巴结病和自身免疫疾病模型和肿瘤异种移植模型中,尿苷与来氟米特的共同给药不会消除来氟米特的免疫抑制和抗肿瘤活性(href=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib38">Xu et al. 1997 , 1999 )。这表明来氟米特通过其他机制发挥其抗增殖和免疫抑制活性(href=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib38">Xu et al. 1997 , 1999 ) )。 我们最近的研究表明,来氟米特和 A77 1726在体外 激酶试验中直接抑制纯化的 S6K1 的活性,并在细胞培养中抑制 S6K1 的活性,IC 50值为 50–75 µM(比其血浆低 4 倍)水平)(=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib11">Doscas et al. 2014)。A77 1726 对 S6K1 活性的抑制导致肿瘤细胞系中 PI-3 激酶途径的反馈激活,这可以通过 AKT 和 S6K1 磷酸化增加以及 S6 磷酸化减少来证明(f=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib11">Doscas等人, 2014 年)。在这里,我们报道 A77 1726 增加 S6K1 和 AKT 磷酸化并刺激 GLUT4 易位至细胞膜和肌管和脂肪细胞中的葡萄糖摄取。 图 1B )。我们进一步表明,来氟米特可控制 ob/ob 小鼠和 HFD 诱导的糖尿病小鼠的高血糖,但不能控制正常小鼠的高血糖。
材料和方法
化学品、抗体和质粒构建体
来氟米特和 A77 1726 由 CinKate Corporation (Oak Park, IL, USA) 友情提供。细胞松弛素和罗格列酮购自 Calbiochem (EMD Millipore)。3-异丁基-1-甲基黄嘌呤 (IBMX)、羧甲基纤维素钠 (CMC)、尿苷、地塞米松和 2-脱氧-葡萄糖 (2-DG) 购自 Sigma Aldrich。2-DG (5–10 Ci (185–370 GBq)/mmol, 1mCi (37 MBq) 购自 PerkinElmer。 体外 研究中使用的胰岛素购自 Invitrogen (Life Technologies)。雷帕霉素(一种 mTOR 抑制剂),针对 PI-3 激酶的 AKT、S6K1、S6、IRS-1、p85 的抗体和磷酸化抗体(AKT S473、AKT T308、S6K1 T389、S6 S235/236、IRS-1 S1101、IR Y1146和 IRS-1 S636)购自 Cell Signaling Technology。抗β-肌动蛋白单克隆抗体购自 Santa Cruz Biotechnology。抗体来源及其应用 见表1 。mCherry-GLUT4-myc 表达载体由 Amira Klip 博士(安大略省多伦多病童医院)友情提供。放射性同位素的使用得到了拉什大学医学中心的批准。所有方法均按照拉什大学医学中心和扬州大学的相关指南和规定进行。
表格1抗体信息。
| 肽/蛋白质靶点 | 抗原序列(如果已知) | 抗体名称 | 制造商、目录编号和/或提供抗体的个人姓名 | 饲养的物种;单克隆或多克隆 | 使用稀释 | RRID(修订版 MS 中需要) |
|---|---|---|---|---|---|---|
| AKT S473 | 未知 | 磷酸-Akt (Ser473)(D9E) Ab | 细胞信号传导 (#4060) | 兔子; 单克隆 | 1:2000 | AB_2315049 |
| AKT T308 | 未知 | 磷酸-Akt (Thr308) 抗体 | 细胞信号 (#9275) | 兔子; 单克隆 | 1:1000 | AB_329828 |
| AKT | 未知 | Akt 抗体 | 细胞信号 (#9272) | 兔子; 单克隆 | 1:1000 | AB_329827 |
| pS6K1 | 未知 | 磷酸化-p70 S6 激酶 (Thr389) (108D2) Ab | 细胞信号 (#9234) | 兔子; 单克隆 | 1:1000 | AB_2269803 |
| S6K1 | 未知 | p70 S6 激酶 (49D7) 抗体 | 细胞信号 (#2708) | 兔子; 单克隆 | 1:1000 | AB_390722 |
| pS6 | 未知 | Phospho-S6 核糖体蛋白 (Ser235/236) (D57.2.2E) Ab | 细胞信号 (#4858) | 兔子; 单克隆 | 1:2000 | AB_916156 |
| S6 | 未知 | S6 核糖体蛋白 (5G10) 抗体 | 细胞信号 (#2217) | 兔子; 单克隆 | 1:1000 | AB_331355 |
| 国税局-1 S1101 | 未知 | Phospho-IRS-1 (Ser1101) 抗体 | 细胞信号 (#2385) | 兔子; 单克隆 | 1:1000 | AB_330363 |
| 国税局-1 S636 | 未知 | Phospho-IRS-1 (Ser636/639) 抗体 | 细胞信号传导 (#2388) | 兔子; 单克隆 | 1:1000 | AB_330339 |
| 国税局-1 | 未知 | IRS-1 (59G8) 抗体 | 细胞信号 (#2390) | 兔子; 单克隆 | 1:1000 | AB_561122 |
| 桥 | 未知 | Phospho-IGF-I Receptor β (Tyr1131)/Insulin Receptor β (Tyr1146) 抗体 | 细胞信号 (#3021) | 兔子; 单克隆 | 1:1000 | AB_331578 |
| 和 | 未知 | 胰岛素受体 β (4B8) 抗体 | 细胞信号传导 (#3025) | 兔子; 单克隆 | 1:1000 | AB_2280448 |
| P85a | 未知 | PI3 激酶 p85 (19H8) 抗体 | 细胞信号传导 (#4257) | 兔子; 单克隆 | WB;1:1000 IP;1:50 | AB_10831521 |
| 肌动蛋白 | 未知 | β-肌动蛋白抗体 (C4) | 圣克鲁斯生物技术 (sc-47778) | 鼠; 单克隆 | 1:200 | AB_626632 |
细胞系和分化
C2C12(鼠成肌细胞系)和 L6 细胞(大鼠成肌细胞系)在补充有 10% 胎牛血清的 DMEM 中培养。对于肌管分化,C2C12细胞的汇合单层在含有10%马血清的DMEM中培养2周,在此期间每两天补充相同的新鲜培养基。对于L6肌管分化,将细胞在含有2%小牛血清的DMEM中培养两周。3T3-L1 脂肪细胞的早期传代(15 代内)根据=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib42">Zebisch等人的详细方案进行了区分。(2012 年)。在无胰岛素培养基中孵育 7-14 天后,超过 95% 的细胞表现出脂肪细胞样表型。所有三种细胞系均购自美国典型培养物保藏中心 (ATCC)。
葡萄糖摄取
C2C12 肌管表达非常低水平的 GLUT4,因此不用于葡萄糖摄取实验。分化的 3T3-L1 脂肪细胞和 L6 肌管在不含胰岛素的 DMEM 培养基中孵育过夜。细胞缺乏血清 4 小时,然后在没有或有 A77 1726 (200 µM) 或雷帕霉素 (20 nM) 的情况下孵育 1 小时,没有或有 2× 氨基酸在必需平衡盐溶液 (EBSS) 中再孵育 1 H。细胞未受刺激或用 20 nM 胰岛素刺激 45 分钟。将未标记的 2-DG (0.1 mM) 和 [ 3 H]-DG 添加到 KRP-HEPES 缓冲液(10 mM HEPES、pH7.4、131.2 mM NaCl、4.7 mM KCl、1.2 mM MgSO 4、2.65 mM )中的细胞中CaCl 2 , 2.5 mM NaH 2 PO 4和 1% 牛血清白蛋白)在 37°C 下 5 分钟。加入 10 µM 细胞松弛素 B 终止反应,然后用冰冷的 PBS 洗涤 3 次。细胞在 0.2 M NaOH 中溶解。在液体闪烁计数器中测量放射性。通过在添加 2-DG 之前将细胞松弛素 B (10 µM) 添加到细胞中来测量非特异性 2-DG 摄取。通过减去非特异性 2-DG 摄取值进行校正后,2-DG 摄取值通过使用 Bio-Rad 蛋白质测定试剂盒 (Bio-Rad) 量化的蛋白质浓度进行标准化。
蛋白质印迹
在 6 孔板中生长的 C2C12 肌管缺乏血清 4 小时。加入 A77 1726 或雷帕霉素并孵育 2 小时。细胞未受刺激或用 20 nM 胰岛素刺激 20 分钟。如葡萄糖摄取实验中所述处理分化的3T3-L1脂肪细胞和L6肌管。在 NP-40 裂解缓冲液(50 mM Tris–HCl (pH 8.0)、150 mM NaCl、1% NP-40、5 mM EDTA、10 µg/mL 抑肽酶、10 µg/mL 亮肽素和 1 mM苯甲基磺酰氟,2 mM 钒酸钠)并用指定的抗体分析蛋白质磷酸化,然后用针对总蛋白质的抗体重新探测。通过使用 NIH ImageJ 软件分析条带的密度,并通过其相应总蛋白质的任意单位进行归一化。sd)来自三个实验( 图 2、3、4和 5 ) 的 条形图 。
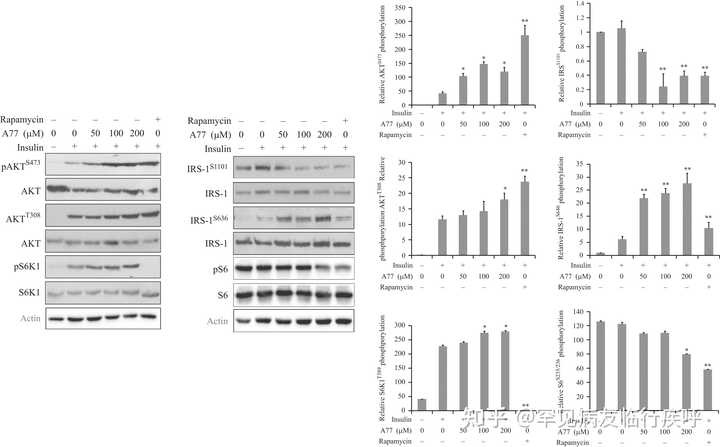

图 2
A77 1726 对 PI-3 激酶途径中蛋白质磷酸化的影响。C2C12 肌管在无血清培养基中饥饿 4 小时,然后用指定浓度的 A77 1726 或雷帕霉素 (50 nM) 处理 2 小时。细胞未受刺激或用胰岛素 (20 nM) 刺激 20 分钟。收获细胞并分析 AKT S473、AKT T308和 S6K1 T389、IRS-1 S1101、IRS-1 S636和 S6 S235/236的磷酸化,然后用它们的特异性抗体重新检测总蛋白水平。通过分析条带的密度确定相对蛋白质磷酸化并以条形图表示。结果是平均值±标准差(sd) 来自三个实验。A77, A77 1726; * P < 0.05;** P < 0.01,与胰岛素刺激对照(无药物治疗)相比。
引文:内分泌学杂志 237, 1; 10.1530/JOE-17-0536
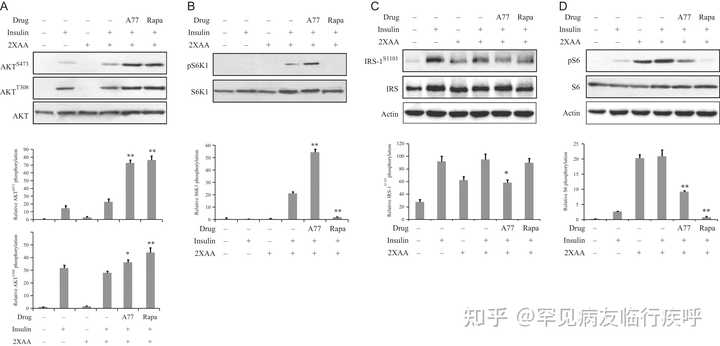

图 3
A77 1726 使 L6 肌管中的胰岛素受体敏感。该实验和其余实验(图 4、5、6 和 7)是在胰岛素抵抗条件下进行的,其中细胞在高浓度氨基酸的存在下孵育。L6 肌管首先缺乏血清 4 小时,然后在不含或存在 A77 1726 的情况下,在不含氨基酸的培养基 (EBSS) 或含有 2 倍氨基酸浓度 (2×AA) 的 EBSS 培养基中培养(200 µM) 2 小时。用胰岛素 (20 nM) 刺激 20 分钟后,收集细胞并分析 AKT S473/T308 (A)、S6K1 T389 (B)、IRS-1 S1101 (C)、S6 S235/236的磷酸化(D),并用它们对总蛋白的特异性抗体进行重新检测。通过使用 Image J 软件分析相对蛋白质磷酸化。结果是三个实验的平均值± 标准差。A77, A77 1726; 拉帕,雷帕霉素。* P < 0.05;** P < 0.01,与对照(使用胰岛素和 2× 氨基酸但未使用药物)相比。
引文:内分泌学杂志 237, 1; 10.1530/JOE-17-0536
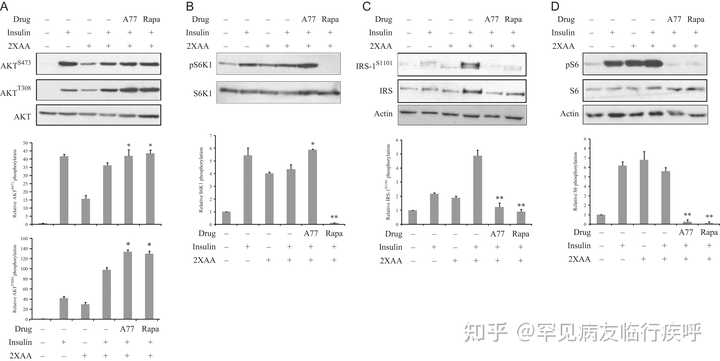

图 4
A77 1726 使 3T3-L1 脂肪细胞中的胰岛素受体敏感。
在图 3
中处理 3T3-L1 脂肪细胞,然后在不含或存在 A77 1726 的情况下,在不含氨基酸的培养基 (EBSS) 或含有 2 倍于 MEM 中发现的氨基酸浓度 (2×AA) 的 EBSS 培养基中孵育2 小时。用胰岛素 (20 nM) 刺激细胞 20 分钟。收获细胞并分析 AKT S473/T308 (A)、S6K1 T389 (B)、IRS-1 S1101 (C)、S6 S235/236 (D) 的磷酸化,并用它们对总蛋白的特异性抗体进行重新检测。通过使用 NIH Image-J 软件分析条带的密度,并通过其相应总蛋白质的任意单位进行归一化。结果是平均值± sd来自三个实验。A77, A77 1726; 拉帕,雷帕霉素。*
P
< 0.05;**
P
< 0.01,与对照(使用胰岛素和 2× 氨基酸但未使用药物)相比。
引文:内分泌学杂志 237, 1;
10.1530/JOE-17-0536
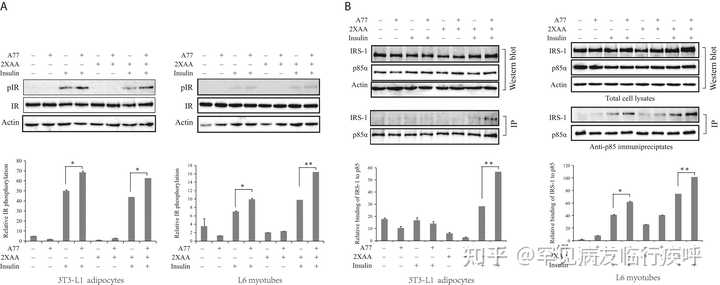

图 5
A77 1726 刺激胰岛素受体酪氨酸磷酸化并增加胰岛素受体底物 (IRS-1) 与 PI-3 激酶的 p85α 亚基的结合。 (A) 3T3-L1 脂肪细胞和 L6 肌管进行了与图 3 类似的处理,并分析了 Y1146 处胰岛素受体酪氨酸磷酸化的磷酸化。(B) A77 1726 增加 PI-3 激酶的 p85 亚基与 IRS-1 的结合。 图3 中处理了3T3-L1脂肪细胞和L6肌管。用抗 p85 抗体对细胞裂解物进行免疫沉淀,然后在蛋白质印迹中用抗 p85 和抗 IRS-1 抗体进行探测。使用Image J软件分析相对蛋白磷酸化。结果是三个实验的平均值± 标准差。A77, A77 1726; * P < 0.05; ** P < 0.01。
引文:内分泌学杂志 237, 1; 10.1530/JOE-17-0536
免疫沉淀
L6 肌管和 3T3-L1 脂肪细胞首先分别被血清饥饿 2 小时或过夜,然后在不含氨基酸的培养基 (EBSS) 或含有 2× 氨基酸 (2×AA) 的 EBSS 培养基中培养,或A77 1726 存在 5 小时。细胞未受刺激或用胰岛素 (100 nM) 刺激 10 分钟。用针对 PI-3 激酶的 p85 亚基的兔单克隆抗体对细胞裂解物进行免疫沉淀,然后在蛋白质印迹中用抗 p85 和抗 IRS1 抗体进行探测。
共聚焦显微镜
按照制造商的方案,使用 FuGENE6 将接种在盖玻片上的未分化 L6 细胞用 mCherry-GLUT4-myc 表达载体 DNA 瞬时转染。孵育 24 小时后,使细胞缺乏血清 2 小时,然后在不存在或存在 A77 1726 (200 µM) 的情况下孵育 4 小时,其中没有或有 EBSS 中氨基酸浓度的 2 倍。细胞未受刺激或用 100 nM 胰岛素刺激 30 分钟。收集盖玻片,在含有 4,6-二脒基-2-苯基吲哚 (DAPI) (0.5 µg/mL; Sigma Chemical) 的 PBS 中用 50% 甘油固定和固定。在 Leica LP8 共聚焦显微镜下观察到 mCherry 标记的 GLUT4 荧光。通过计算每次处理中随机选择的 10 个区域,计算总 mCherry-GLUT4 表达细胞中 GLUT4 易位进入质膜的阳性细胞百分比。结果代表平均值±标准偏差(sd ) 来自具有相似结果的三个实验之一。
动物和药物管理
动物的使用和所有实验方案均经拉什大学医学中心机构动物护理和使用委员会和扬州大学兽医学院批准。所有小鼠都保持 12 小时的光照/黑暗循环,并在 23°C 的环境温度下饲养在通风的笼子中。雄性
ob/ob
小鼠 (B6.V-Lep ob /OlaHsd,雄性) 购自 Harlan Laboratories, Inc.。这些小鼠在正常食物饮食 (NCD) 上
随意喂食。
CMC(1.5%溶解在蒸馏水中)用作制备来氟米特的载体。小鼠(8-10 周龄)通过管饲法给予 1.5% CMC 或来氟米特。尿苷与来氟米特联合给予
ob/ob
小鼠来确定当各种组织中的嘧啶核苷酸水平正常化时,来氟米特是否仍能控制高血糖。尿苷剂量基于我们之前的研究 ( href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib38">Xu et al. 1997 ,
1999
),即每天两次 2 g/kg 足以使快速增殖的肿瘤细胞或
lpr/lpr
小鼠淋巴细胞中的嘧啶核苷酸水平正常化或超调。通过腹膜内(ip)注射给予溶解在盐水中的尿苷。
ob/ob
用载体(每天 1.5% CMC)、来氟米特(35 mg/kg/天,每天,灌胃)、尿苷(2 g/kg,每天两次,ip)或来氟米特(35 mg/kg/天,每天)治疗小鼠, 灌胃) + 尿苷 (2 g/kg, 每天两次, ip) 三天。禁食 6 小时后,从尾静脉采血并通过拜耳手持式血糖仪进行分析。在治疗前和治疗后3天测量小鼠的体重。处理后第 3 天在各个笼子中测量食物摄入量。
对于葡萄糖耐量试验(GTT)和胰岛素耐量试验(ITT),小鼠如上处理3天。第 4 天,小鼠禁食并在 GTT 前 6 小时用最后一剂治疗。通过腹膜内注射用葡萄糖(1 g/kg)攻击小鼠。对于 ITT,对小鼠进行类似处理并禁食 6 小时,然后将胰岛素(2.5 单位/千克)静脉注射到尾静脉。通过使用拜耳血糖仪在 0 到 120 分钟 (GTT) 或 0 到 60 分钟 (ITT) 之间的不同时间点测量血糖水平。使用 GraphPad Prism 5 软件计算 GTT 和 ITT 中葡萄糖水平的 AUC(曲线下面积)。AUC 绘制为条形图。
C57BL/6 雄性小鼠购自扬州大学比较医学中心。给小鼠(5 周大)喂食 NCD 或 HFD(按重量计 24% 脂肪、24% 蛋白质、41% 碳水化合物、1% 其他,这转化为卡路里百分比为 20% 蛋白质、35% 碳水化合物、45% 脂肪)(江苏医药有限公司,扬州,中国)为期 10 周。如前所述,用 CMC 或来氟米特处理小鼠,然后用 GTT。喂食 NCD 或 HFD 的 C57BL/6 小鼠中的 ITT 与上述类似,只是腹膜内注射胰岛素 (2.5 单位/公斤)。
体内 AKT 激活
ob/ob 小鼠每天治疗三天。他们在第 4 天给予最后一剂,然后禁食 6 小时。注射胰岛素(2.5 单位/公斤,静脉注射)5 分钟后,处死小鼠。收集腓肠肌、肠系膜内脏白色脂肪和肝组织(50-100 mg/样品)并立即在 NP-40 裂解缓冲液中均质化。使用 Pierce BCA 蛋白质测定试剂盒(Thermo Fisher Scientific)测量蛋白质浓度。通过蛋白质印迹分析AKT S473/T308、S6 S235/236和 IRS-1 S1101磷酸化。通过使用 NIH ImageJ 软件分析条带的密度,并通过其相应总蛋白质的任意单位进行归一化。
统计分析
数据表示为平均值±标准偏差 ( sd )(葡萄糖摄取和 GLUT4 易位测定)或平均值的标准误差 ( sem )(血糖水平)。未配对的学生 t 检验用于分析不同组中 3T3-L1 脂肪细胞和 L6 肌管中葡萄糖摄取的差异,以及 ImageJ 分析中任意数量的蛋白质印迹数据的差异。通过重复测量方差分析(方差分析)分析不同治疗组之间的血糖水平差异。 通过配对Student t 检验统计分析治疗前后各组血糖水平的差异。一个 _ 值 <0.05 被认为具有统计学意义。所有统计数据均使用 SigmaPlot 11 软件(Systat Software,San Jose,CA,USA)进行。
结果
A77 1726 诱导 PI-3 激酶通路的反馈激活
A77 1726 在肿瘤细胞系中对 S6K1 的抑制导致通过胰岛素样生长因子 1 (IGF-1) 受体反馈激活 PI-3 激酶通路 ( =" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib11">Doscas et al. 2014 )。在这里,我们测试了 A77 1726 是否也诱导 C2C12 肌管中 PI-3 激酶通路的反馈激活。如图 2 所示,胰岛素诱导C2C12肌管中IRS-1 S636、AKT T308、AKT S473和S6K1 T389的磷酸化。A77 1726 增强胰岛素诱导的 AKT S473、AKT T308和 S6K1 T389的磷酸化,但抑制 S6 S235/236和 IRS的磷酸化S1101,两者都被S6K1磷酸化。相反,A77 1726 增加了 IRS S636的磷酸化( 图 2 )。该位点主要被 mTOR 磷酸化。值得注意的是,S6 S235/236和 IRS-1 S1101在未刺激的 C2C12 肌管中高度磷酸化。mTOR 抑制剂雷帕霉素增加 AKT T308和 AKT S473磷酸化,但抑制 IRS S1101、IRS S636、S6 S235/236和 S6K1 T389磷酸化。A77 1726 增加的 S6K1 磷酸化与先前对 PF-4708671 的观察结果一致,PF-4708671 是一种特异性 S6K1 抑制剂(f=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib23">Pearce等。2010 年,< a h ref=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib30">沉等人。2016 年)。
A77 1726 增强胰岛素受体信号
我们接下来检查了 A77 1726 对 L6 肌管和 3T3-L1 脂肪细胞中 PI-3 激酶途径中蛋白质磷酸化的影响,在高氨基酸浓度的情况下,胰岛素抵抗的情况。
如图3
所示,在L6肌管中不存在氨基酸的情况下,胰岛素诱导AKT S473/T308磷酸化。与先前的观察结果一致 ( f="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib22">Patti et al. 1998 ),高氨基酸激活 S6K1,如增加的 S6 S235/236和 IRS-1 S1101 所示磷酸化,这被认为是胰岛素抵抗的间接证据。A77 1726 诱导 S6K1 的磷酸化,S6K1 是 mTOR 的下游分子,而雷帕霉素抑制它。A77 1726 和雷帕霉素均抑制 IRS-1 S1101和 S6 S235/236磷酸化,但增加L6 肌管中的AKT S473和 AKT T308磷酸化。在 3T3-L1 脂肪细胞中获得了类似的结果(
图 4
)。
<
a
href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib34">嗯等人。(2004) 报道说,与野生型小鼠相比,S6K1 缺乏会导致胰岛素受体致敏,这可以通过胰岛素治疗小鼠肝脏中的胰岛素受体酪氨酸磷酸化增加来证明。在这里,我们测试了 A77 1726 是否确实可以增强胰岛素受体信号。3T3-L1 脂肪细胞和 L6 肌管在不存在或存在 2× 氨基酸的情况下与 A77 1726 预孵育,并保持未刺激或用胰岛素刺激 30 分钟。如图 5A 所示,在不存在或存在高浓度氨基酸的情况下培养的 3T3-L1 脂肪细胞和 L6 肌管中胰岛素诱导的胰岛素受体酪氨酸磷酸化。A77 1726 进一步增加了这种磷酸化。免疫沉淀显示胰岛素刺激增加了 PI-3 激酶的 p85 亚基与在不存在或存在 2× 氨基酸的情况下培养的 3T3-L1 脂肪细胞和 L6 肌管中的 IRS-1 的结合。A77 1726 (
图 5B
)。通过蛋白质印迹分析细胞裂解物。在用于免疫沉淀的细胞裂解物中发现了相等水平的 IRS-1 和 PI-3 激酶的 p85 亚基(
图 5B
)。
A77 1726 增加 GLUT4 易位至细胞膜
AKT 激活诱导 GLUT4 从囊泡转移到质膜中(ref=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib9">Dann等人2007 年, Copps & White 2012 年 )。在未刺激的 L6 的细胞质中检测到 GLUT4,但在没有氨基酸的情况下在胰岛素刺激的细胞中转移到质膜( 图 6) )。在存在 2 个氨基酸的情况下,单独的 A77 1726 略微增加了 L6 细胞中的 GLUT4 膜易位。在 2× 氨基酸存在的情况下,胰岛素也弱诱导 GLUT4 易位到质膜,几乎与没有 2× 氨基酸的情况一样,这可能是由于在该实验中使用了未分化的 L6 成肌细胞。或者,在高功率场下相对较低的转染效率和少数 GLUT4 阳性细胞可能会屏蔽 2x 氨基酸介导的 GLUT4 易位抑制。然而,A77 1726 在 2 倍氨基酸浓度的存在下显着增加了 L6 细胞中胰岛素刺激的 GLUT4 膜易位( 图 6 )。
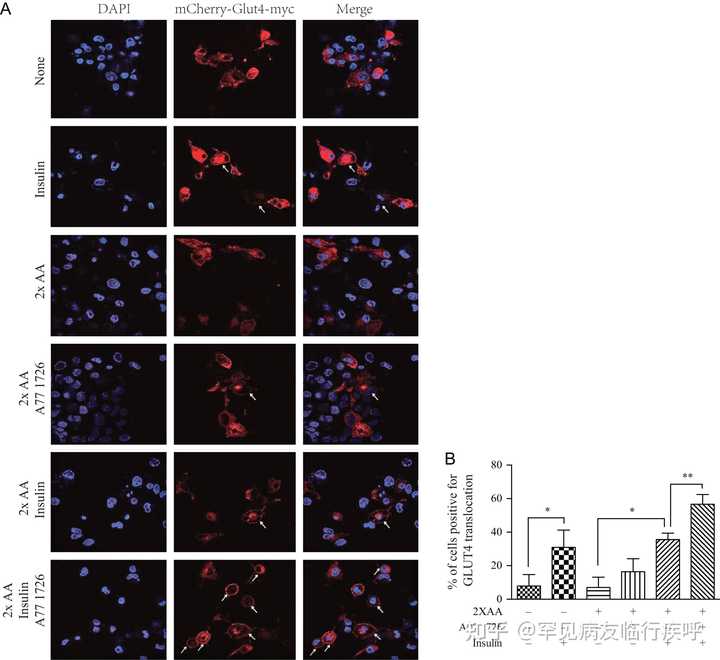

图 6
A77 1726 增强胰岛素诱导的 GLUT4 易位至质膜。(A) GLUT4 易位至质膜。mCherry-GLUT4-myc 转染的 L6 细胞缺乏血清 4 小时,然后在没有或有 2 倍氨基酸浓度的 A77 1726 (200 µM) 存在或不存在的情况下孵育。细胞未受刺激或用 20 nM 胰岛素刺激 45 分钟。在甲醇中固定 10 分钟后,在 Leica LP8 共聚焦显微镜下观察到 mCherry 标记的 GLUT4 荧光。箭头表示 mCherry 标记的 GLUT4 易位至细胞膜。(B) GLUT4 易位到质膜的量化。数据代表具有相似结果的三个实验之一的平均值± 标准差。* P < 0.05;** 磷 < 0.01。此图的全彩版本可在 https:// doi.org/10.1530/JOE-17- 0536 获得。
引文:内分泌学杂志 237, 1; 10.1530/JOE-17-0536
A77 1726 增加葡萄糖摄取
我们接下来确定 A77 1726 对胰岛素受体的敏化是否导致葡萄糖摄取增加。用高浓度氨基酸处理的细胞被认为处于胰岛素抵抗状态。事实上,与相应的对照相比,2 倍的氨基酸浓度显着降低了 L6 肌管中葡萄糖摄取的基础水平 29%,并将胰岛素刺激的葡萄糖摄取降低了 22%( 图 7A )。A77 1726 在存在 2 倍氨基酸浓度的情况下将胰岛素刺激的葡萄糖摄取增加了 31%。作为阳性对照的雷帕霉素增加了 19% 的葡萄糖摄取( 图 7A ) 在存在 2 倍氨基酸浓度的情况下,在胰岛素刺激的 L6 肌管中。用 3T3-L1 脂肪细胞观察到 A77 1726 对胰岛素诱导的葡萄糖摄取的稍微更好的刺激( 图 7B )。值得注意的是,A77 1726 是一种细胞抑制药物,不会影响分化的非分裂细胞的活力和增殖。细胞仅在 A77 1726 存在下孵育几个小时。使用增强型细胞计数 Kit-8 (CCK-8),我们发现 A77 1726 (200 µM) 对 3T3-L1 脂肪细胞和 L6 肌管没有任何细胞毒性(数据未显示)。使用 CellTiter-Glo Luminescent Cell Viability Assay,我们发现 A77 1726 (200 µM) 在孵育 48 小时后将 3T3-L1 脂肪细胞的增殖降低了约 20%(数据未显示)。
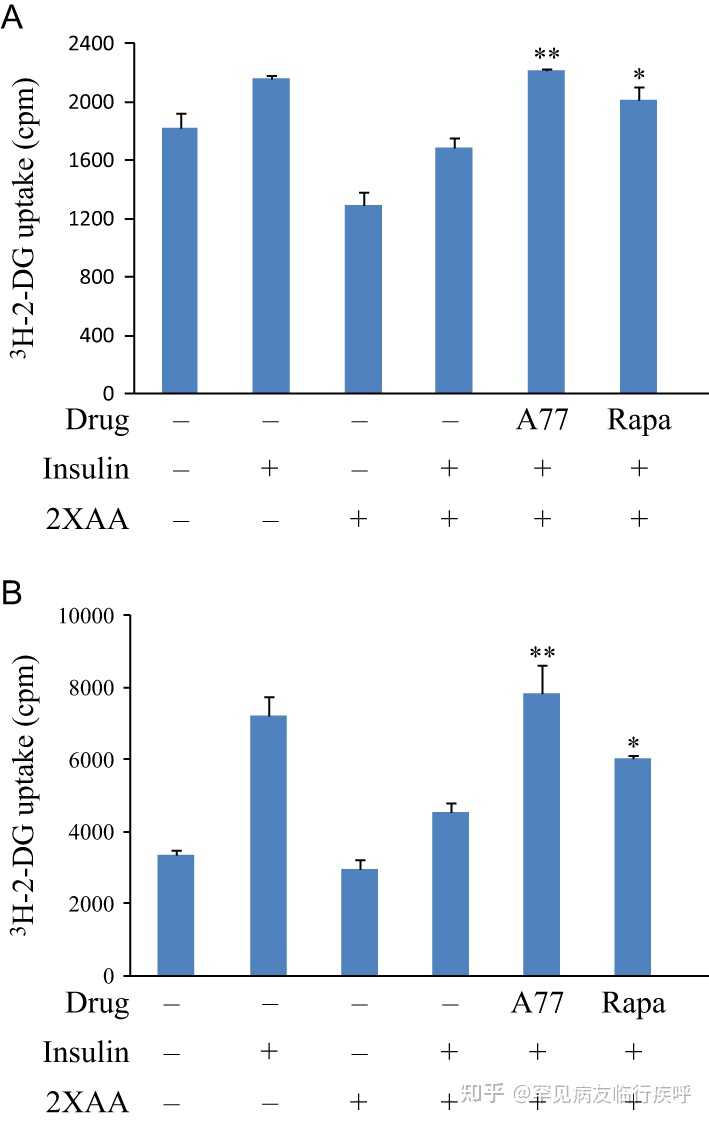

图 7
A77 1726 增加葡萄糖摄取。接种在 24 孔板中的 L6 肌管 (A) 和 3T3-L1 脂肪细胞 (B) 在无血清、低葡萄糖 DMEM 培养基中饥饿 4 小时。然后将细胞在 EBSS 中在不存在或存在 2×AA 和/或 A77 1726 (200 µM) 或雷帕霉素 (50 nM) 的情况下孵育 2 小时。细胞未受刺激或用 20 nM 胰岛素刺激 45 分钟,然后将 [ 3 H]-2-DG 温育 5 分钟。数据是 具有相似结果的三个实验之一中一式三份的平均值±标准差。* P < 0.05;** P < 0.01,与对照(使用胰岛素和 2× 氨基酸但未使用药物)相比。
引文:内分泌学杂志 237, 1; 10.1530/JOE-17-0536
来氟米特控制高血糖
我们首先评估了来氟米特控制雄性小鼠高血糖的能力,因为雌性小鼠体内的雌激素可以预防高脂饮食诱导的代谢综合征。雌性动物容易发生脂肪组织储存和葡萄糖稳态,而雄性动物则易患糖尿病。如 表 2所示,治疗前 ob/ob 小鼠的血糖水平非常高(>200 mg/dL),而用 CMC(一种用于溶解来氟米特的载体)治疗的小鼠的血糖水平仍然非常高。在单独使用来氟米特或来氟米特加尿苷(<110 mg/dL)治疗的小鼠中,空腹血糖水平降至正常水平。统计分析显示,来氟米特治疗显着降低血糖水平( P < 0.01)。 用尿苷治疗,一种用于使体外 和 体内 嘧啶核苷酸水平正常化的核苷(href=" https:// joe.bioscientifica.com/ configurable/content/journals $002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib38">Xu et al. 1997 , 1999 ),对空腹血糖水平没有影响,也没有阻断来氟米特介导的高血糖控制。这些观察结果表明,来氟米特控制高血糖症与其对嘧啶核苷酸合成的抑制作用无关。用 CMC、来氟米特、尿苷或来氟米特加尿苷治疗 3 天的小鼠在食物摄入和体重方面没有显着差异( 表 2 )。
表 2来氟米特对 ob/ob 小鼠高血糖的控制。†
| 药品 | 小鼠数量(N) | 血糖浓度 (mg/dL) | 体重(克) | 食物摄入量(克/天) | P值* | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 前 | 后 | P值 | 前 | 后 | 改变 | P值* | ||||
| 车辆 | 6 | 234 ± 16 | 252 ± 22 | 0.697 | 47.8 ± 2.9 | 48.0 ± 3.1 | 0.12 ± 0.21 | 4.9 ± 0.23 | ||
| 来氟米特 | 7 | 275 ± 12 | 111 ± 6 | 0.001 | 48.4 ± 0.48 | 48.5 ± 0.51 | −0.02 ± 0.13 | 0.252 | 5.0 ± 0.09 | 0.687 |
| 尿苷 | 4 | 210 ± 12 | 253 ± 15 | 0.153 | 48.7 ± 0.68 | 48.5 ± 0.69 | −0.02 ± 0.08 | 0.793 | 4.9 ± 0.07 | 1.000 |
| Lef + 尿苷 | 4 | 222 ± 15 | 97 ± 15 | 0.021 | 48.5 ± 1.21 | 48.4 ± 1.06 | −0.3 ± 0.04 | 0.385 | 4.8 ± 0.04 | 0.272 |
†
ob/ob
小鼠(雄性,8-10 周)用载体(1.5% CMC,每天,管饲)、来氟米特(35 mg/kg,每天,管饲)、尿苷(2 g/kg,每天两次, ip)或来氟米特(35 mg/kg,每天,管饲)+尿苷(2 g/kg,每天两次,ip),持续三天。禁食6小时后,测量血糖水平并通过配对Student
t
检验进行统计学分析。在治疗前和治疗后3天测量小鼠的体重。在治疗后第 3 天,在单独的笼子中测量个体小鼠的食物消耗。
*
与车辆控制相比。
我们接下来进行 GTT 以检查来氟米特降低ob/ob
小鼠血糖水平的能力。接受来氟米特或来氟米特加尿苷的小鼠的血糖水平显着低于用CMC治疗的小鼠(
P
<0.001)(
图8A
)。单独的尿苷治疗并没有显着改变小鼠的血糖水平。ITT测定显示,在注射胰岛素15分钟后,对照小鼠或用尿苷处理的小鼠的血糖水平升高(
图8C
)。相比之下,胰岛素不会增加来氟米特治疗小鼠的血糖水平,但能够略微增加用尿苷加来氟米特治疗的小鼠的血糖水平(
图 8C )
)。
将胰岛素注射到对照ob/ob
小鼠尾部后血糖水平的瞬时升高可能是由压力或胰岛素受体内化引起的。注射胰岛素后血糖水平的短暂升高与其他人的观察结果一致(href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib17">He et al. 2013)。与单独使用 CMC 或尿苷治疗的小鼠相比,来氟米特或来氟米特加尿苷治疗的小鼠的 AUC 显着降低(
图 8B
和
D
)(
P
< 0.01)。
一致地,来氟米特治疗显着降低 GTT(图 8E
和
F
)和 ITT 测定(
图 8G
)中的血糖水平和
H
) 在 HFD 喂养的小鼠中,与用 CMC 治疗的小鼠相比。Leflunomide 在降低
ob/ob
小鼠的血糖水平方面似乎更有效,而在 HFD 诱导的糖尿病小鼠中仅适度降低血糖水平。来氟米特治疗没有显着改变喂食 NCD 的小鼠的血糖水平(
图 8E
、
F
、
G
和
H
)。
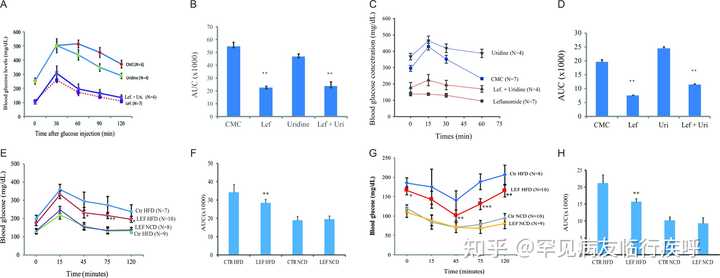

图 8
来氟米特控制高血糖和胰岛素受体敏化。(A) ob/ob 小鼠按照“材料和方法”部分所述进行治疗。通过腹膜内注射对小鼠进行 GTT 葡萄糖 (1 g/kg) 攻击。在指定时间测量血糖水平。CMC vs 来氟米特、尿苷或来氟米特+尿苷, P < 0.001;来氟米特 vs 来氟米特 + 尿苷, P = 0.002。左,来氟米特;Uri,尿苷;计算 AUC 的任意值并在条形图 (B) 中显示 **CMC 与来氟米特或来氟米特 + 尿苷, P < 0.001;CMC 与尿苷, P = 0.057。(C) ITT 中来氟米特对高血糖的控制。 ob/ob 如“材料和方法”部分所述对小鼠进行处理。禁食6小时后,静脉注射胰岛素(2.5 U/kg)。在指定时间测量血糖水平。CMC vs 来氟米特、尿苷或来氟米特+尿苷, P < 0.001;来氟米特 vs 来氟米特 + 尿苷, P = 0.006。(D) 不同治疗组的 AUC 显示为条形图。**CMC 对比来氟米特或来氟米特 + 尿苷, P < 0.001;CMC 与尿苷, P = 0.115。(E 和 G) 每天用 CMC 或来氟米特 (35 mg/kg/天) 治疗以正常食物 (NCD) 或高脂肪饮食 (HFD) 喂养 10 周的 C57BL/6 雄性小鼠 (5 周龄) ) 3 天,然后评估 GTT (E) 和 ITT (G) 中的血糖水平。(F 和 H) 不同治疗组的 AUC 显示为条形图。** CMC 与来氟米特在喂食 HFD 的小鼠中, P < 0.001;在喂食 NCD 的小鼠中,CMC 与来氟米特相比, P > 0.05。此图的全彩版本可在 https:// doi.org/10.1530/JOE-17- 0536 获得。
引文:内分泌学杂志 237, 1; 10.1530/JOE-17-0536
- 下载图
-
将图下载为 PowerPoint 幻灯片
最后,我们测试了来氟米特治疗是否增强了 ob/ob 小鼠 体内 的胰岛素受体信号传导。 如图9A 所示,与未刺激的对照组相比,胰岛素显着增加了正常小鼠肌肉、脂肪和肝脏中AKT S473/T308的磷酸化( P <0.05)( 图9C )。在对照小鼠的这些胰岛素敏感组织中,来氟米特治疗没有或仅略微增强胰岛素诱导的 AKT 磷酸化。对照组和来氟米特治疗小鼠组织中AKT S473/T308磷酸化水平相当( 图 9A 和 C )( P > 0.05)。相比之下,胰岛素在 ob/ob 小鼠的肌肉、脂肪和肝脏中对AKT S473/T308磷酸化的诱导效果较差( 图9B )。光密度分析显示,胰岛素敏感组织中的 AKT S473/T308磷酸化在未处理和胰岛素处理的小鼠之间没有显着差异( P > 0.05)( 图 9D )。与仅用对照载体治疗的小鼠相比,来氟米特治疗显着增加了肌肉和脂肪组织中胰岛素刺激的 AKT S473/T308磷酸化 ( P < 0.05)。AKT S473/T308 来氟米特治疗的ob/ob 小鼠肝脏中的磷酸化水平似乎高于未治疗的 ob/ob 小鼠。然而,统计分析没有达到显着性。 同样,用来氟米特加尿苷处理的ob/ob 小鼠的肌肉、脂肪和肝脏中的AKT S473/T308磷酸化水平也高于单独用尿苷处理的那些(补充图 1,见最后给出的 补充数据 部分)本文)。然而,未处理和来氟米特处理的小鼠的肌肉、脂肪和肝脏中的 S6 和 IRS-1 磷酸化没有显着差异(补充图 2)。
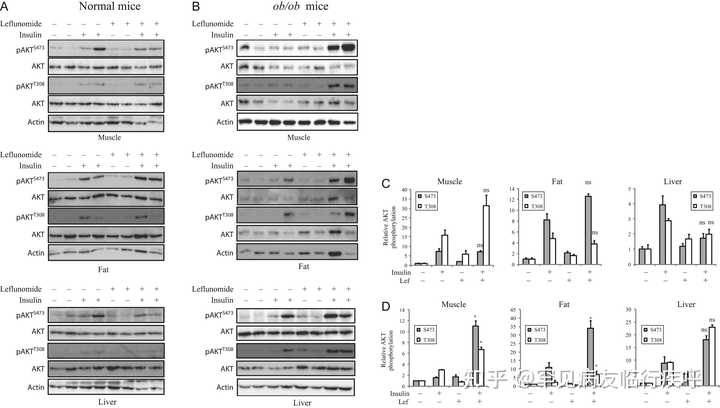

图 9
来氟米特可增强代谢组织中的 AKT 磷酸化。C57BL/6 (A) 和 ob/ob (B) 小鼠每天用 CMC (1.5%) 或来氟米特 (35 mg/kg/天) 治疗 3 天。第 4 天,小鼠接受最后一剂治疗,然后禁食 6 小时。然后给小鼠静脉注射盐水或胰岛素(2.5 单位/千克)。五分钟后,处死小鼠。收获腓肠肌、内脏白色脂肪和肝组织,并用它们的特异性抗体通过蛋白质印迹分析 AKTS473/T308磷酸化,并用抗总 AKT 蛋白的抗体重新探测。免疫印迹代表每组两只动物的数据。该实验重复两次。(C 和 D) 正常 C57BL/6 小鼠组织中的条带密度 (C) 和 ob/ob 小鼠 (D) 使用 NIH Image-J 软件进行分析,并通过其相应总蛋白的任意单位进行标准化。结果是 每组四只动物的平均值±标准差。* P < 0.05;ns,在 CMC/胰岛素治疗组和来氟米特/胰岛素治疗组之间不显着。
引自:内分泌学杂志237,1; 10.1530/JOE-17-0536
讨论
S6K1 过度激活与胰岛素抵抗以及高血糖和肥胖症的发展有关( href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib34">Um等人, 2004 年,
2006 年
)。S6K1 缺陷型小鼠对 HFD 诱导的高血糖和肥胖具有抵抗力( href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib34">Um等人, 2004 年,
2006 年
)。 href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib30">舒姆等人。(2016)最近报道,PF-4708671 是一种 S6K1 抑制剂,通过使胰岛素受体敏感来控制 HFD 喂养小鼠的高血糖症。我们最近将 S6K1 鉴定为来氟米特和 A77 1726 的分子靶标 ( ="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib11">Doscas et al. 2014)。A77 1726 在 A375 黑色素瘤细胞中对 S6K1 活性的抑制导致 PI-3 激酶通路的反馈激活,这可以通过增加 AKT S473和减少 S6 S235/236磷酸化来证明。我们目前的研究表明,在正常和/或胰岛素抵抗条件下,A77 1726 对 S6K1 活性的抑制导致小鼠 C2C12 和大鼠 L6 肌管以及小鼠 3T3-L1 脂肪细胞中 S6 S235/236和 IRS-1 S1101磷酸化的抑制。A77 1726 增强胰岛素诱导的胰岛素受体酪氨酸磷酸化,PI-3 激酶的 p85 亚基与 IRS-1 的结合(
图 5
),以及在高氨基酸浓度存在的情况下,胰岛素诱导的 GLUT4 易位至 L6 细胞的质膜(
图 6
)。来氟米特治疗增加
体内
肌肉和脂肪组织中的AKT T308/S473磷酸化
(图 9B)
。这些观察结果共同表明 A77 1726 对 S6K1 的抑制是其抗高血糖作用的主要原因。
郭
等人
。
(1997 年
) 早些时候报道,长期使用来氟米特对健康大鼠的血糖水平没有影响。由于正常动物的胰岛素敏感组织没有胰岛素抵抗,来氟米特不能进一步使胰岛素受体敏感。一致地,我们发现用 CMC 或来氟米特处理的 NCD 喂养的瘦小鼠的血糖水平没有显着差异(
图 8E
、
F
、
G
和
H
)。
我们研究了 A77 1726 对 S6K1、S6 S235/236和 IRS-1 S1101的两种相关底物磷酸化的抑制作用。由于 PI-3 激酶通路的反馈激活,A77 1726 处理的细胞中 S6K1 磷酸化增加。A77 1726
在体外
抑制肌管和脂肪细胞中的 S6 S235/236和 IRS-1 S1101磷酸化。相比之下,来氟米特治疗并未显着降低
体内
胰岛素刺激小鼠代谢组织中的S6 S235/236和 IRS-1 S1101(补充图 2)。与我们的观察类似,(
Shum
et al
. 2016
) 报道了 PF-4708671,一种比 A77 1726 更有效的 S6K1 抑制剂,在体内
不抑制 S6 磷酸化。这些研究人员认为,代偿性 S6K2 激活可能会屏蔽 PF-4708671 对 S6 磷酸化的抑制作用(ref="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib30">Shum等人, 2016 年)。href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib23">皮尔斯等人。(2010)据报道,在血清存在下,PF-4708671 不抑制 293 细胞中的早期 S6 磷酸化。我们推测 A77 1726 或 PF-4708671 对 S6K1 活性的抑制可能最初会抑制 S6 和 IRS-1 磷酸化。然而,当 S6K1 被进一步反馈激活时,S6 活性的不完全抑制可能是 S6 和 IRS-1 磷酸化抑制不足的原因。在胰岛素注射后几分钟内,泄漏的 S6K1 活性足以使 S6 和 IRS-1 完全磷酸化。然而,一旦 AKT 被反馈激活,AKT 磷酸化水平将在一段时间内保持高水平。活化的 AKT 继续调节葡萄糖代谢,直到它被去磷酸化和失活。IRS-1 在 S636 被 mTOR 磷酸化(
Copps & White 2012
)。我们目前的研究表明,由于 mTOR 的反馈激活,A77 1726在 C2C12 肌管中诱导 IRS-1 S636磷酸化。有趣的是,ROCK1 对 IRS-1 S636的磷酸化导致 PI-3 激酶的激活 (
Copps & White 2012
)。因此,A77 1726 可通过抑制 IRS-1 S1101和增加 IRS-1 S636磷酸化来改善胰岛素受体信号传导。
来氟米特和 A77 1726 抑制 S6K1 活性,IC 50值约为 50–75 µM ( ="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib11">Doscas et al. 2014 )。使用来氟米特(20 mg/天)治疗的 RA 患者的血浆 A77 1726 浓度高于 200 µM(ref="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib5">Chan等人, 2005 年)。用 35 mg/kg 剂量的来氟米特处理的小鼠血液中的 A77 1726 的半衰期非常长,为 15 小时。A77 1726 的血液浓度在 4 小时内达到峰值 500 µM,并在小鼠单剂量 35 mg/kg 来氟米特后 24 小时保持在 250 µM ( f="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib6">Chong et al. 1999 ))。这些数据表明血浆中的 A77 1726 浓度高到足以抑制 S6K1 活性。事实上,来氟米特治疗小鼠的代谢组织中 AKT 磷酸化增加。用 35 mg/kg/天剂量的来氟米特治疗
ob/ob
小鼠的血糖水平降至正常水平。与尿苷合用不能阻断来氟米特的抗血糖作用。一项较早的临床研究表明,接受来氟米特治疗的 RA 患者血液中的平均葡萄糖水平 (83 mg/dL) 显着低于接受其他方案治疗的患者 (93 mg/dL) ( ref="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib25">Rho et al. 2009)。大约 10% 接受来氟米特治疗的患者体重减轻不能归因于腹泻或其他胃肠道副作用,可能是由于代谢率增加(f="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib7">Coblyn等人, 2001 年)。这些观察结果共同表明 A77 1726 浓度足以抑制 S6K1 活性,从而更好地控制高血糖。炎性细胞因子,特别是 TNF-α,也可以使胰岛素受体脱敏并导致高血糖症的发展(
Copps & White 2012
)。抗 TNF-α 治疗不会降低 RA 患者的血糖水平 (
Rosenvinge et al. 2007
)。与来氟米特不同,许多抗炎药不会降低血糖水平(href="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib25">Rho等人, 2009 年),这表明抑制炎症并不是更好地控制高血糖的充分解释。
我们意识到我们研究中的几个弱点。
首先,支持来氟米特通过抑制体内
S6K1 的活性使胰岛素受体敏感并控制高血糖这一结论的证据很弱。来氟米特治疗的胰岛素抵抗小鼠组织中增加的 AKT 磷酸化间接表明由于 S6K1 活性的抑制,PI-3 激酶通路的反馈激活。然而,在来氟米特治疗的动物组织中未观察到 S6 和 IRS-1 磷酸化的抑制作用。这与使用 PF-4708671(一种有效的 S6K1 抑制剂)的研究中的观察结果一致(ref="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib30">Shum等人, 2016 年)。
其次,来氟米特在降低ob/ob
中的血糖水平方面效果非常好小鼠,即使样本量相对较小,
P
值也达到了统计学显着水平。然而,在 HFD 诱导的糖尿病小鼠模型中,来氟米特仅适度降低 GTT 和 ITT 中的血糖水平。
第三,来氟米特治疗显着增强了ob/ob
小鼠肌肉和脂肪组织中胰岛素刺激的 AKT 磷酸化。来氟米特治疗增加了
ob/ob肝脏中的 AKT 磷酸化
小鼠,但由于动物数量相对较少和 AKT 磷酸化的高度可变性,这在统计学上并不显着。第四,由于已经有许多抗糖尿病药物可用于治疗高血糖,来氟米特可能仅对特定类型的患者群体有用,例如糖尿病和类风湿关节炎患者。我们研究中使用的糖尿病小鼠模型没有 RA,因此不能完全概括认为使用来氟米特的临床环境。
长期使用雷帕霉素并不能控制小鼠模型和患者的高血糖症,反而会加剧高血糖症(
"https://joe.bioscientifica.com/configurable/content/journals$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib10">Di Paolo等人2006 年
,
Schindler等人2014 年
)。在长期使用后,雷帕霉素未能控制高血糖是因为它能够抑制负责磷酸化 AKT S473的 mTORC2 的活性(
图 1B
) (
Lamming
等人,
2012 年
)。在本研究中,我们没有跟踪长期使用来氟米特后的降血糖作用。然而,有几条证据表明长期使用来氟米特可能会保持其治疗效果:(1)与雷帕霉素不同,A77 1726 抑制 S6K1(ref="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib11">多斯卡斯等人。2014),mTOR下游的激酶。因此,A77 1726 不像雷帕霉素那样抑制 mTORC2(
图 1B
);(2) PF-4708671 长期使用后的抗高血糖作用
在体内
是可持续的( ef="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib30">Shum et al. 2016 )。A77 1726 和 PF-4708671 均通过抑制 S6K1 来控制高血糖;(3) 长期使用来氟米特治疗的 RA 患者的血糖水平明显低于使用其他药物治疗的患者 ( ref="
https://
joe.bioscientifica.com/
configurable/content/journals
$002fjoe$002f237$002f1$002fJOE-17-0536.xml?t:ac=journals%24002fjoe%24002f237%24002f1%24002fJOE-17-0536.xml#bib25">Rho et al. 2009 )。尽管如此,来氟米特在长期使用后能否保持持久的治疗效果,应首先在动物模型中验证,然后才能在患者中进行研究。
总之,我们目前的研究表明,A77 1726 通过抑制 S6K1 活性诱导 PI-3 激酶途径的反馈激活,并且 A77 1726在体外
增加葡萄糖摄取和 GLUT4 易位至质膜(
图 1B
)。A77 1726 还可以通过刺激糖原合成和通过激活的 AKT 抑制糖异生来改善葡萄糖代谢(
图 1B
)。来氟米特能够控制高血糖并改善
体内
胰岛素敏感性。来氟米特可能对治疗同时患有 2 型糖尿病的 RA 患者特别有用。
补充数据
这链接到 https:// doi.org/10.1530/JOE-17- 0536 上的论文在线版本。
利益申报
作者声明不存在可能被视为损害所报告研究的公正性的利益冲突。
资金
这项工作得到了 Rush Pilot 糖尿病研究基金、中国自然科学基金 (81672463) 和江苏省高等教育机构优先学术项目发展对 X Xu 的资助,以及对 Y Li 的 NIH R01 (CA204926) 资助。
作者贡献声明
XX 构思概念,撰写手稿并研究数据。杰西; JS,医学,JY;AJW 进行了实验并参与了讨论;YL(芝加哥大学)参与讨论;YL(贝勒医学院)RAP 参与了讨论、审阅和编辑手稿。
致谢
作者非常感谢 CinKate 公司的 James W. Williams 博士为 mCherry-GLUT4-myc 表达载体提供 A77 1726 和来氟米特以及 Amira Klip 博士(安大略省多伦多病童医院)。
参考
-
Boura-Halfon S & Zick Y 2009IRS 蛋白的磷酸化、胰岛素作用和胰岛素抵抗。
美国生理学-内分泌与代谢杂志
296E581–E591。(
https://
doi.org/10.1152/ajpendo
.90437.2008
)
-
Breedveld FC 和Dayer JM 2000来氟米特:治疗类风湿性关节炎的作用方式。
风湿病年鉴
59841–849。(
https://
doi.org/10.1136/ard.59.
11.841
)
-
Bruneau JM , Yea CM , Spinella - Jaegle S , Fudali C , Woodward K , Robson PA , Sautes C , Westwood R , Kuo EA , Williamson RA ,等人。1998人二氢乳清酸脱氢酶的纯化及其被来氟米特的活性代谢物 A77 1726 抑制。
生化杂志
336299–303。(
https://
doi.org/10.1042/bj33602
99
)
-
Cannon GW & Kremer JM 2004来氟米特。
北美风湿病诊所
30295–309。(
https://
doi.org/10.1016/j.rdc.2
004.01.010
)
-
Chan V , Charles BG & Tett SE 2005类风湿性关节炎患者服用来氟米特后的人群药代动力学和 A77 1726 血浆浓度与疾病活动度测量之间的关联。
英国临床药理学杂志
60257–264。(
https://
doi.org/10.1111/j.1365-
2125.2005.02415.x
)
-
Chong AS , Huang W , Liu W , Luo J , Shen J , Xu W , Ma L , Blinder L , Xiao F , Xu X ,et al. 1999来氟米特的体内活性:药代动力学分析和免疫抑制机制。
移植
68100–109。(
https://
doi.org/10.1097/0000789
0-199907150-00020
)
-
Coblyn JS 、 Shadick N 和Helfgott S 2001来氟米特在类风湿性关节炎中的相关体重减轻。
关节炎和风湿病学
441048–1051。(
https://
doi.org/10.1002/1529-01
31(200105)
44:5<1048::AID-ANR184>3.0.CO;2-V)
-
Copps KD & White MF 2012通过胰岛素受体底物蛋白 IRS1 和 IRS2 的丝氨酸/苏氨酸磷酸化调节胰岛素敏感性。
糖尿病学
552565–2582。(
https://
doi.org/10.1007/s00125-
012-2644-8
)
-
Dann SG 、 Selvaraj A 和Thomas G 2007mTOR Complex1-S6K1 信号:处于肥胖、糖尿病和癌症的十字路口。
分子医学趋势
13252–259。(
https://
doi.org/10.1016/j.molme
d.2007.04.002
)
-
Di Paolo S 、 Teutonico A 、 Leogrande D 、 Capobianco C 和Schena PF 2006慢性抑制哺乳动物雷帕霉素靶点信号下调胰岛素受体底物 1 和 2 以及 AKT 激活:癌症和糖尿病之间的十字路口?
美国肾脏病学会杂志
172236–2244。(
https://
doi.org/10.1681/ASN.200
6030196
)
-
Doscas ME 、 Williamson AJ 、 Usha L 、 Bogachkov Y 、 Rao GS 、 Xiao F 、 Wang Y 、 Ruby C 、 Kaufman H 、 Zhou J 等。2014A77 1726 对 p70 S6 激酶 (S6K1) 活性的抑制及其对细胞增殖和细胞周期进程的影响。
瘤形成
16824–834。(
https://
doi.org/10.1016/j.neo.2
014.08.006
)
-
Elder RT , Xu X , Williams JW , Gong H , Finnegan A & Chong AS 1997来氟米特的免疫抑制代谢物 A77 1726 通过两种生化机制影响小鼠 T 细胞。
免疫学杂志
15922-27。
-
Fenton TR & Gout IT 201070kDa 核糖体 S6 激酶的功能和调节。
国际生物化学和细胞生物学杂志
4347-59。(
https://
doi.org/10.1016/j.bioce
l.2010.09.018
)
-
郭S 2013胰岛素抵抗的分子基础:IRS和Foxo1在控制糖尿病及其并发症中的作用。
今日药物发现:疾病机制
10e27–e33。(
https://
doi.org/10.1016/j.ddmec
.2013.06.003
)
-
Guo S 2014胰岛素信号传导、耐药性和代谢综合征:从小鼠模型到疾病机制的见解。
内分泌学杂志
220T1–T23。(
https://
doi.org/10.1530/JOE-13-
0327
)
-
Guo Z , Chong AS , Shen J , Foster P , Sankary HN , McChesney L , Mital D , Jensik SC , Gebel H & Williams JW 1997来氟米特对正常胰岛和同基因胰岛移植功能的体内影响。
移植
63716–721。(
https://
doi.org/10.1097/0000789
0-199703150-00018
)
-
He J , Gao J , Xu M , Ren S , Stefanovic-Racic M , O'Doherty RM & Xie W 2013PXR 消融缓解小鼠的饮食诱导和遗传性肥胖和胰岛素抵抗。
糖尿病
621876–1887 年。(
https://
doi.org/10.2337/db12-10
39
)
-
Herlitz-Cifuentes HS , Garces PC , Fernandez LI & Guzman-Gutierrez EA 2015全身炎症对类风湿性关节炎胰岛素和葡萄糖代谢功能的影响。
当前的糖尿病评论
12156–162。(
https://
doi.org/10.2174/1573399
811666150602150325
)
-
江平、李海 、李 新, 2015 年类风湿性关节炎中的糖尿病危险因素:系统评价和荟萃分析。
临床和实验风湿病学
33115–121。
-
Lamming DW 、 Ye L 、 Katajisto P 、 Goncalves MD 、 Saitoh M 、 Stevens DM 、 Davis JG 、 Salmon AB 、 Richardson A 、 Ahima RS 等。2012雷帕霉素诱导的胰岛素抵抗是由 mTORC2 丢失介导的,并且与长寿无关。
科学
3351638-1643。(
https://
doi.org/10.1126/science
.1215135
)
-
Nathan DM 2015糖尿病:诊断和治疗进展。
美国医学会杂志
3141052-1062。(
https://
doi.org/10.1001/jama.20
15.9536
)
-
Patti ME , Brambilla E , Luzi L , Landaker EJ & Kahn CR 1998氨基酸对胰岛素作用的双向调节。
临床研究杂志
1011519–1529。(
https://
doi.org/10.1172/JCI1326
)
-
Pearce LR 、 Alton GR 、 Richter DT 、 Kath JC 、 Lingardo L 、 Chapman J 、 Hwang C 和Alessi DR 2010PF-4708671 的表征,一种新型且高度特异性的 p70 核糖体 S6 激酶 (S6K1) 抑制剂。
生化杂志
431245–255。(
https://
doi.org/10.1042/BJ20101
024
)
-
Pinto AJ , Roschel H , de Sa Pinto AL , Lima FR , Pereira RMR , Silva CA , Bonfa E & Gualano B 2017和久坐行为:自身免疫性风湿性疾病中被忽视的危险因素?
自身免疫审查
16667–674。(
https://
doi.org/10.1016/j.autre
v.2017.05.001
)
-
Rho YH 、 Oeser A 、 Chung CP 、 Milne GL 和Stein CM 2009用于治疗类风湿性关节炎的药物:当前使用与心血管危险因素之间的关系。
药物信息档案
234–40。(
https://
doi.org/10.1111/j.1753-
5174.2009.00019.x
)
-
Rosenvinge A 、 Krogh-Madsen R 、 Baslund B 和Pedersen BK 2007类风湿性关节炎患者的胰岛素抵抗:抗 TNFα 治疗的效果。
斯堪的纳维亚风湿病学杂志
3691–96。(
https://
doi.org/10.1080/0300974
0601179605
)
-
Ruckemann K 、 Fairbanks LD 、 Carrey EA 、 Hawrylowicz CM 、 Richards DF 、 Kirschbaum B 和Simmonds HA 1998来氟米特抑制健康人有丝分裂原刺激的 T 淋巴细胞中的嘧啶从头合成。
生物化学杂志
27321682-21691。(
https://
doi.org/10.1074/jbc.273
.34.21682
)
-
Schindler CE , Partap U , Patchen BK & Swap SJ 2014慢性雷帕霉素治疗导致雄性小鼠患糖尿病。
美国生理学杂志:调节、综合和比较生理学
307R434–R443。(
https://
doi.org/10.1152/ajpregu
.00123.2014
)
-
Selman C 、 Tullet JM 、 Wieser D 、 Irvine E 、 Lingard SJ 、 Choudhury AI 、 Claret M 、 Al-Qassab H 、 Carmignac D 、 Ramadani F 等。2009核糖体蛋白 S6 激酶 1 信号调节哺乳动物的寿命。
科学
326140-144。(
https://doi.org/10.1126/science.1177221
)
-
Shum M 、 Bellmann K 、 St-Pierre P 和Marette A 2016S6K1 的药理抑制可增加体外和饮食诱导的肥胖小鼠的葡萄糖代谢和 Akt 信号传导。
糖尿病学
59592–603。(
https://
doi.org/10.1007/s00125-
015-3839-6
)
- Siemasko KF 、 Chong AS 、 Williams JW 、 Bremer EG 和Finnegan A 1996免疫抑制剂来氟米特对 B 细胞功能的调节。 移植 61635–642。( https:// doi.org/10.1097/0000789 0-199602270-00020 )
- Siemasko K 、 Chong AS 、 Jack HM 、 Gong H 、 Williams JW 和Finnegan A 1998免疫抑制药物来氟米特抑制 JAK3 和 STAT6 酪氨酸磷酸化导致 IgG1 产生受阻。 免疫学杂志 1601581–1588。
- 嗯SH 、 Frigerio F 、 Watanabe M 、 Picard F 、华金M 、贴纸M 、 Fumagalli S 、 Allegrini PR 、 Kozma SC 、 Auwerx J 等。2004S6K1 的缺失可防止年龄和饮食引起的肥胖,同时增强胰岛素敏感性。 自然 431200–205。( https:// doi.org/10.1038/nature0 2866 )
- Um SH 、 D'Alessio D 和Thomas G 2006 年营养过剩、胰岛素抵抗和核糖体蛋白 S6 激酶 1、S6K1。 细胞代谢 3393–402。( https:// doi.org/10.1016/j.cmet. 2006.05.003 )
- Versini M 、 Jeandel PY 、 Rosenthal E 和Shoenfeld Y 2014自身免疫性疾病中的肥胖:不是被动的旁观者。 自身免疫评论 13981–1000。( https:// doi.org/10.1016/j.autre v.2014.07.001 )
- Williamson RA 、 Yea CM 、 Robson PA 、 Curnock AP 、 Gadher S 、 Hambleton AB 、 Woodward K 、 Bruneau JM 、 Hambleton P 、 Spinella-Jaegle S 等。1996二氢乳清酸脱氢酶是来氟米特生物效应的靶点。 移植诉讼 283088–3091。
- Xia T , Cheng Y , Zhang Q , Xiao F , Liu B , Chen S & Guo F 2012中枢神经系统中的 S6K1 通过 MC4R/CRH 通路调节能量消耗,以应对必需氨基酸的剥夺。 糖尿病 612461–2471。( https:// doi.org/10.2337/db11-12 78 )
- Xu X , Williams JW , Bremer EG , Finnegan A & Chong AS 1995新型免疫抑制剂来氟米特抑制 T 细胞中蛋白质酪氨酸磷酸化。 生物化学杂志 27012398-12403。( https:// doi.org/10.1074/jbc.270 .21.12398 )
- Xu X , Williams JW , Gong H , Finnegan A & Chong AS 1996来氟米特免疫抑制代谢物的两种活性,A77 1726。抑制嘧啶核苷酸合成和蛋白质酪氨酸磷酸化。 生化药理学 52527-534。( https:// doi.org/10.1016/0006-29 52(96)00303-6 )
- Xu X , Blinder L , Shen J , Gong H , Finnegan A , Williams JW & Chong AS 1997来氟米特控制 MRL/MpJ-lpr/lpr 小鼠淋巴组织增生和自身免疫性疾病的体内机制。 免疫学杂志 159167–174。
- Xu X , Shen J , Mall JW , Myers JA , Huang W , Blinder L , Saclarides TJ , Williams JW & Chong AS 1999新型免疫调节药物来氟米特的体外和体内抗肿瘤活性:作用机制。 生化药理学 581405-1413。( https:// doi.org/10.1016/S0006-2 952(99)00228-2 )
- Zebisch K 、 Voigt V 、 Wabitsch M 和Brandsch M 2012协议,用于将 3T3-L1 细胞有效分化为脂肪细胞。 分析生物化学 42588-90。( https:// doi.org/10.1016/j.ab.20 12.03.005 )
- Zimmet P 、 Alberti KG 和Shaw J 2001 年糖尿病流行的全球和社会影响。 自然 414782-787。( https:// doi.org/10.1038/414782a )
- Zimmet PZ 、 Magliano DJ 、 Herman WH 和Shaw JE 2014糖尿病:21 世纪的挑战。 柳叶刀糖尿病和内分泌学 256-64。( https:// doi.org/10.1016/S2213-8 587(13)70112-8 )
| 以上为谷歌翻译,未经校对存在大量错误,仅供专人人士校正用。版权归原作者。 |
|---|
英文原文:
Control of hyperglycemia in male mice by leflunomide: mechanisms of action in Journal of Endocrinology
Authors: Junhong Chen 1 , 2 , Jing Sun 1 , 2 , Michelle E Doscas 3 , Jin Ye 3 , Ashley J Williamson 4 , Yanchun Li 5 , Yi Li 6 , Richard A Prinz 7 , and Xiulong Xu 1 , 2 , 3 , 8
- 1 Institute of Comparative Medicine, Yangzhou University, Yangzhou, Jiangsu Province, China
- | 2 College of Veterinary Medicine, Yangzhou University, Yangzhou, Jiangsu Province, China
- | 3 Department of Cell and Molecular Medicine, Rush University Medical Center, Chicago, Illinois, USA
- | 4 Rush Medical College, Rush University Medical Center, Chicago, Illinois, USA
- | 5 Section of Endocrinology, Department of Medicine, University of Chicago, Chicago, Illinois, USA
- | 6 Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, Texas, USA
- | 7 Department of Surgery, NorthShore University Health System, Evanston, Illinois, USA
- | 8 Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonosis, Yangzhou University, Yangzhou, China
DOI:
https://
doi.org/10.1530/JOE-17-
0536
Volume/Issue:
Volume 237: Issue 1
Page Range: 43–58
Article Type: Research ArticleOnline Publication Date: Apr 2018Copyright: © 2018 Society for Endocrinology 2018
Abstract
p70 S6 kinase (S6K1) is a serine/threonine kinase that phosphorylates the insulin receptor substrate-1 (IRS-1) at serine 1101 and desensitizes insulin receptor signaling. S6K1 hyperactivation due to overnutrition leads to hyperglycemia and type 2 diabetes. Our recent study showed that A77 1726, the active metabolite of the anti-rheumatoid arthritis (RA) drug leflunomide, is an inhibitor of S6K1. Whether leflunomide can control hyperglycemia and sensitize the insulin receptor has not been tested. Here we report that A77 1726 increased AKTS473/T308 and S6K1T389 phosphorylation but decreased S6S235/236 and IRS-1S1101 phosphorylation in 3T3-L1 adipocytes, C2C12 and L6 myotubes. A77 1726 increased insulin receptor tyrosine phosphorylation and binding of the p85 subunit of the PI-3 kinase to IRS-1. A77 1726 enhanced insulin-stimulated glucose uptake in L6 myotubes and 3T3-L1 adipocytes, and enhanced insulin-stimulated glucose transporter type 4 (GLUT4) translocation to the plasma membrane of L6 cells. Finally, we investigated the anti-hyperglycemic effect of leflunomide on ob/ob and high-fat diet (HFD)-induced diabetes mouse models. Leflunomide treatment normalized blood glucose levels and overcame insulin resistance in glucose and insulin tolerance tests in ob/ob and HFD-fed mice but had no effect on mice fed a normal chow diet (NCD). Leflunomide treatment increased AKTS473/T308 phosphorylation in the fat and muscle of ob/ob mice but not in normal mice. Our results suggest that leflunomide sensitizes the insulin receptor by inhibiting S6K1 activity in vitro , and that leflunomide could be potentially useful for treating patients with both RA and diabetes.
Keywords: leflunomide ; p70 S6 kinase ; insulin resistance ; insulin receptor substrate ; hyperglycemia
Introduction
Type 2 diabetes is a major public health problem (="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib43
">Zimmet et al. 2001,
2014
). Although many anti-diabetic medications are available, some have intolerable side-effects or lose their therapeutic efficacy after the long-term use (
Nathan 2015
). Failure to control hyperglycemia leads to diabetic complications, which account for most diabetes-related morbidity and mortality (
Nathan 2015
). Because of physical inactivity and circulating inflammatory cytokines, diabetic patients with rheumatoid arthritis (RA) may have worse hyperglycemia than those without RA (
Herlitz-Cifuentes et al. 2015
, f="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib24
">Pinto et al. 2017). Individuals with RA have a significantly higher risk of developing type 2 diabetes (f="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib19
">Jiang et al. 2015) and obesity (
"https://joe.bioscientifica.com/view/journals/joe/237/1/JOE-17-0536.xml#bib35">Versini et al. 2014
) than those without RA. Currently, patients with both RA and diabetes are treated with antidiabetic and anti-RA drugs separately. A drug that controls both RA and hyperglycemia could greatly benefit patients with both problems.
The binding of insulin to its receptor activates the insulin receptor tyrosine kinase, leading to insulin receptor autophosphorylation and the phosphorylation of intracellular protein substrates such as the insulin receptor substrates (IRS) (
Fig. 1B
) (
Guo 2013
,
2014
). Tyrosine-phosphorylated IRS interact with the p85 subunit of the PI-3 kinase and activate its catalytic p110 subunit (
Fig. 1B
). PI-3 kinase activation leads to serine phosphorylation and activation of the protein kinase B (AKT) (
Guo 2013
,
2014
). AKT activation plays a critical role in glucose metabolism (ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib9
">Dann et al. 2007,
Copps & White 2012
). AKT activation stimulates glucose uptake by inducing translocation of the glucose transporter type 4 (GLUT4) to the plasma membrane of both adipose and muscle cells (
Fig. 1
) (
Guo 2013
,
2014
). In addition, AKT also regulates glucose metabolism by stimulating glycogen synthesis and inhibiting gluconeogenesis (
Fig. 1B
) (
Guo 2013
,
2014
).
Figure 1
The mechanisms of leflunomide-mediated anti-hyperglycemic effect. (A) Chemical structure of leflunomide and A77 1726. (B) Mode of action of A77 1726. Overnutrition with high concentrations of fatty acids and amino acids leads to constitutive S6K1 activation, which phosphorylates IRS-1S1101, leading to poor AKT activation. Leflunomide and its active metabolite A77 1726 inhibit S6K1 activity, subsequently leading to the inhibition of IRS-1S1101. Inhibition of IRS-1S1101 phosphorylation leads to insulin receptor sensitization, as revealed by increased insulin receptor tyrosine phosphorylation and increased binding of IRS-1 to the p85 subunit of PI-3 kinase. AKTS473/T308 phosphorylation and activation leads to increased glucose uptake by stimulating GLUT4 membrane translocation, increased glycogen synthesis, and decreased gluconeogenesis. Chronic use of rapamycin leads to inhibition of both mTORC1 and mTORC2, thus exacerbating hyperglycemia. A full colour version of this figure is available at https:// doi.org/10.1530/JOE-17- 0536 .
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
- Download Figure
-
Download figure as PowerPoint slide
The mechanistic target of rapamycin (mTOR) kinase is a serine/threonine kinase activated by AKT. Overnutrition with high concentrations of amino acids and fatty acids also activates mTOR ( Fig. 1B ). p70 S6 kinase (S6K1), a serine/threonine protein kinase downstream of mTOR, phosphorylates the IRS and subsequently attenuates the activation of the PI3K pathway ( Fig. 1B ) ( Fenton & Gout 2010 ). Constitutive S6K1 activation by hyperinsulinemia or overnutrition leads to insulin receptor desensitization ( Boura-Halfon & Zick 2009 , Copps & White 2012 ). S6K1 is also involved in regulating the expression of several energy expenditure-related genes such as the melanocortin-4 receptor (MC4R) (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib34 ">Um et al. 2004, 2006 , ref=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib37 ">Xia et al. 2012). S6K1−/− mice do not develop obesity and hyperglycemia when fed a high-fat diet (HFD) (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib34 ">Um et al. 2004). These mice have a significantly longer life span than the wild-type mice (=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib29 ">Selman et al. 2009). Insulin receptor signaling is highly active in the metabolic tissues of the HFD-fed S6K1−/− mice, as evidenced by increased AKT phosphorylation in their liver, muscle and fat (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib34 ">Um et al. 2004). S6K1 is a key kinase driving insulin resistance and inducing obesity under conditions of nutrient overload (ef=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib9 ">Dann et al. 2007).
Leflunomide is an orally administered prodrug proscribed for treating rheumatoid arthritis (RA) ( Breedveld & Dayer 2000 ). Upon indigestion, it is rapidly and completely (>99%) converted in the gastrointestinal tract, plasma and liver to its active metabolite, A77 1726 ( Fig. 1A ) ( Breedveld & Dayer 2000 ). Once in plasma, A77 1726 is avidly bound to plasma proteins, mainly albumin ( Cannon & Kremer 2004 ). A77 1726 has a very long half-life of 15.5 days (range 14–18 days) and is cleared after it is metabolized into trifluoromethylaniline-oxanilic acid (60–70%) and excreted into the urine ( Breedveld & Dayer 2000 , Cannon & Kremer 2004 ). A77 1726 is the only active metabolite of leflunomide and is solely responsible for its therapeutic activity. A77 1726 inhibits the activity of protein tyrosine kinases and dihydroorotate dehydrogenase (DHO-DHase) (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib40 ">Xu et al. 1995, 1996 , 1997 , 1999 , Siemasko et al. 1996 , 1998 , f=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib12 ">Elder et al. 1997, Ruckemann et al. 1998 ). The ability of A77 1726 to inhibit DHO-DHase activity (the IC50 values of approximately 100 nM) is about 10–100 times stronger than its ability to inhibit the activity of protein tyrosine kinases such as p56lck, p59fyn and the PDGF receptor (the IC50 values of approximately 25–50 µM) (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib40 ">Xu et al. 1995, 1996 , 1997 , 1999 , Ruckemann et al. 1998 ). Inhibition of pyrimidine nucleotide synthesis by leflunomide was thought to be its primary mechanism of action ( Williamson et al. 1996 , "https://joe.bioscientifica.com/view/journals/joe/237/1/JOE-17-0536.xml#bib3">Bruneau et al. 1998 ). However, addition of exogenous uridine, which normalizes pyrimidine nucleotide levels in vitro in cell cultures, only partially antagonizes this anti-proliferative effect. Co-administration of uridine with leflunomide in a lymphadenopathy and autoimmune disease model of MRL/MpJ- lpr/lpr mice and in a tumor xenograft model does not abrogate the immunosuppressive and antitumor activities of leflunomide (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib38 ">Xu et al. 1997, 1999 ). This suggests that leflunomide exerts its anti-proliferative and immunosuppressive activity by other mechanisms (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib38 ">Xu et al. 1997, 1999 ). Our recent study revealed that leflunomide and A77 1726 directly inhibit the activity of purified S6K1 in an in vitro kinase assay and inhibit the activity of S6K1 in cell culture, with an IC50 value of 50–75 µM (4-fold lower than its plasma levels in patients) (=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib11 ">Doscas et al. 2014). Inhibition of S6K1 activity by A77 1726 leads to feedback activation of the PI-3 kinase pathway in tumor cell lines, as evidenced by increased AKT and S6K1 phosphorylation and decreased S6 phosphorylation (=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib11 ">Doscas et al. 2014). Here, we report that A77 1726 increases S6K1 and AKT phosphorylation and stimulates GLUT4 translocation to the cell membrane and glucose uptake in myotubes and adipocytes ( Fig. 1B ). We further show that leflunomide controls hyperglycemia in ob/ob mice and in the mice with HFD-induced diabetes but not in normal mice.
Materials and methods
Chemicals, antibodies and plasmid construct
Leflunomide and A77 1726 were kindly provided by CinKate Corporation (Oak Park, IL, USA). Cytochalasin and rosiglitazone were purchased from Calbiochem (EMD Millipore). 3-isobutyl-1-methylxanthine (IBMX), carboxymethyl-cellulose sodium (CMC), uridine, dexamethasone and 2-deoxy-glucose (2-DG) were purchased from Sigma Aldrich. 2-DG (5–10 Ci (185–370 GBq)/mmol, 1mCi (37 MBq) was purchased from PerkinElmer. Insulin used in the
in vitro
study was purchased from Invitrogen (Life Technologies). Rapamycin (an mTOR inhibitor), antibodies against AKT, S6K1, S6, IRS-1, p85 of the PI-3 kinase and phospho-antibodies (AKTS473, AKTT308, S6K1T389, S6S235/236, IRS-1S1101, IRY1146 and IRS-1S636) were purchased from Cell Signaling Technology. Anti-β-actin monoclonal antibody was purchased from Santa Cruz Biotechnology. The sources of antibodies and their applications were listed in
Table 1
. mCherry-GLUT4-myc expression vector was kindly provided by Dr Amira Klip (The Hospital for Sick Children, Toronto, Ontario). Use of the radioactive isotope was approved by Rush University Medical Center. All methods were performed in accordance with the relevant guidelines and regulations of Rush University Medical Center and Yangzhou University.
Table 1Antibodies information.
| Peptide/protein target | Antigen sequence (if known) | Name of Antibody | Manufacturer, catalog #, and/or name of individual providing the antibody | Species raised in; monoclonal or polyclonal | Dilution used | RRID (required in revised MSs) |
|---|---|---|---|---|---|---|
| AKTS473 | Unknown | Phospho-Akt (Ser473)(D9E) Ab | Cell Signaling (#4060) | Rabbit; monoclonal | 1:2000 | AB_2315049 |
| AKTT308 | Unknown | Phospho-Akt (Thr308) Ab | Cell Signaling (#9275) | Rabbit; monoclonal | 1:1000 | AB_329828 |
| AKT | Unknown | Akt Antibody | Cell Signaling (#9272) | Rabbit; monoclonal | 1:1000 | AB_329827 |
| pS6K1 | Unknown | Phospho-p70 S6 Kinase (Thr389) (108D2) Ab | Cell Signaling (#9234) | Rabbit; monoclonal | 1:1000 | AB_2269803 |
| S6K1 | Unknown | p70 S6 Kinase (49D7) Ab | Cell Signaling (#2708) | Rabbit; monoclonal | 1:1000 | AB_390722 |
| pS6 | Unknown | Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) Ab | Cell Signaling (#4858) | Rabbit; monoclonal | 1:2000 | AB_916156 |
| S6 | Unknown | S6 Ribosomal Protein (5G10) Ab | Cell Signaling (#2217) | Rabbit; monoclonal | 1:1000 | AB_331355 |
| IRS-1S1101 | Unknown | Phospho-IRS-1 (Ser1101) Antibody | Cell Signaling (#2385) | Rabbit; monoclonal | 1:1000 | AB_330363 |
| IRS-1S636 | Unknown | Phospho-IRS-1 (Ser636/639) Antibody | Cell Signaling (#2388) | Rabbit; monoclonal | 1:1000 | AB_330339 |
| IRS-1 | Unknown | IRS-1 (59G8) Ab | Cell Signaling (#2390) | Rabbit; monoclonal | 1:1000 | AB_561122 |
| pIR | Unknown | Phospho-IGF-I Receptor β (Tyr1131)/Insulin Receptor β (Tyr1146) Antibody | Cell Signaling (#3021) | Rabbit; monoclonal | 1:1000 | AB_331578 |
| IR | Unknown | Insulin Receptor β (4B8) Ab | Cell Signaling (#3025) | Rabbit; monoclonal | 1:1000 | AB_2280448 |
| P85α | Unknown | PI3 Kinase p85 (19H8) Ab | Cell Signaling (#4257) | Rabbit; monoclonal | WB;1:1000 IP;1:50 | AB_10831521 |
| Actin | Unknown | β-Actin Antibody (C4) | Santa Cruz Biotechnology (sc-47778) | Mouse; monoclonal | 1:200 | AB_626632 |
Cell lines and differentiation
C2C12 (a murine myoblast cell line) and L6 cells (a rat myoblast cell line) were cultured in DMEM supplemented with 10% fetal bovine serum. For myotube differentiation, the confluent monolayers of C2C12 cells were cultured in DMEM containing 10% horse serum for 2 weeks during which the same fresh medium was replenished every two days. For L6 myotube differentiation, the cells were cultured in DMEM containing 2% calf serum for two weeks. The early passages of 3T3-L1 adipocytes (within 15 passages) were differentiated according to a detailed protocol of
"https://joe.bioscientifica.com/view/journals/joe/237/1/JOE-17-0536.xml#bib42">Zebisch et al. (2012
). After incubation in insulin-free medium for 7–14 days, more than >95% of the cells exhibited an adipocyte-like phenotype. All three cell lines were purchased from the American Type Culture Collection (ATCC).
Glucose uptake
C2C12 myotubes express very low levels of GLUT4 and therefore were not used for glucose uptake experiments. Differentiated 3T3-L1 adipocytes and L6 myotubes were incubated overnight in insulin-free DMEM medium. Cells were starved of serum for 4 h and then incubated in the absence or presence of A77 1726 (200 µM) or rapamycin (20 nM) for 1 h without or with 2× amino acids in essential balanced salt solution (EBSS) for another 1 h. Cells were left unstimulated or stimulated with 20 nM insulin for 45 min. Unlabeled 2-DG (0.1 mM) and [3H]-DG were added to the cells in the KRP-HEPES buffer (10 mM HEPES, pH7.4, 131.2 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 2.65 mM CaCl2, 2.5 mM NaH2PO4, and 1% bovine serum albumin) at 37°C for 5 min. The reaction was terminated by adding 10 µM cytochalasin B followed by wash with ice-cold PBS three times. The cells were lysed in 0.2 M NaOH. The radioactivity was measured in a liquid scintillation counter. Nonspecific 2-DG uptake was measured by adding cytochalasin B (10 µM) into the cells prior to the addition of 2-DG. The values of 2-DG uptake, after correction by subtracting the value of the non-specific 2-DG uptake, were normalized by protein concentrations, which were quantified by using a Bio-Rad Protein Assay kit (Bio-Rad).
Western blot
C2C12 myotubes grown in 6-well plates were starved of serum for 4 h. A77 1726 or rapamycin was added and incubated for 2 h. Cells were left unstimulated or stimulated with 20 nM insulin for 20 min. Differentiated 3T3-L1 adipocytes and L6 myotubes were treated as described in the glucose uptake experiments. Cell lysates were prepared in NP-40 lysis buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 5 mM EDTA, 10 µg/mL aprotinin, 10 µg/mL leupeptin, and 1 mM phenylmethylsulfonyl fluoride, 2 mM sodium vanadate) and analyzed for protein phosphorylation with the indicated antibodies, followed by reprobing with antibodies against total proteins. The density of the bands was analyzed by using NIH ImageJ software and normalized by the arbitrary units of their corresponding total proteins. Quantified results were presented as the mean ± standard deviation (s.d.) from three experiments (
Figs 2
,
3
,
4
and
5
) in bar graphs.
Figure 2
Effect of A77 1726 on protein phosphorylation in the PI-3 kinase pathway. C2C12 myotubes were starved in serum-free medium for 4 h and then treated with the indicated concentrations of A77 1726 or rapamycin (50 nM) for 2 h. The cells were left unstimulated or stimulated with insulin (20 nM) for 20 min. Cells were harvested and analyzed for the phosphorylation of AKTS473, AKTT308, and S6K1T389, IRS-1S1101, IRS-1S636, and S6S235/236, followed by reprobing with their specific antibodies for total protein levels. Relative protein phosphorylation was determined by analyzing the density of bands and presented as bar graphs. The results are the mean ± standard deviation (s.d.) from three experiments. A77, A77 1726; * P < 0.05; ** P < 0.01, compared to the insulin-stimulated control (no drug treatment).
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
Figure 3
A77 1726 sensitizes insulin receptor in L6 myotubes. This experiment and the remaining ones (Figs 4, 5, 6 and 7) were carried out under the condition of insulin resistance in which the cells were incubated in the presence of high concentrations of amino acids. L6 myotubes were first starved of serum for 4 h and then incubated either in an amino-acid free medium (EBSS) or EBSS medium containing 2× the amino acid concentrations (2×AA) found in MEM in the absence or presence of A77 1726 (200 µM) for 2 h. After stimulation with insulin (20 nM) for 20 min, cells are harvested and analyzed for the phosphorylation of AKTS473/T308 (A), S6K1T389 (B), IRS-1S1101 (C), S6S235/236 (D), and reprobed with their specific antibodies for total proteins. Relative protein phosphorylation was analyzed by using an Image J software. The results are the mean ± s.d. from three experiments. A77, A77 1726; Rapa, rapamycin. * P < 0.05; ** P < 0.01, compared to the control (with insulin and 2× amino acids but without drug).
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
Figure 4
A77 1726 sensitizes insulin receptor in 3T3-L1 adipocytes. 3T3-L1 adipocytes were treated in Fig. 3 and then incubated either in an amino-acid free medium (EBSS) or EBSS medium containing 2× the amino acid concentrations (2×AA) found in MEM in the absence or presence of A77 1726 for 2 h. Cells were stimulated with insulin (20 nM) for 20 min. The cells were harvested and analyzed for the phosphorylation of AKTS473/T308 (A), S6K1T389 (B), IRS-1S1101 (C), S6S235/236 (D), and reprobed with their specific antibodies for total proteins. The density of bands was analyzed by using an NIH Image-J software and normalized by the arbitrary units of their corresponding total proteins. The results are the mean ± s.d. from three experiments. A77, A77 1726; Rapa, rapamycin. * P < 0.05; ** P < 0.01, compared to the control (with insulin and 2× amino acids but without drug).
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
Figure 5
A77 1726 stimulates insulin receptor tyrosine phosphorylation and increases the insulin receptor substrate (IRS-1) binding to the p85α subunit of the PI-3 kinase. (A) 3T3-L1 adipocytes and L6 myotubes were similarly treated as in Fig. 3 and analyzed for the phosphorylation of insulin receptor tyrosine phosphorylation at Y1146. (B) A77 1726 increases the binding of the p85 subunit of the PI-3 kinase to IRS-1. 3T3-L1 adipocytes and L6 myotubes were treated in Fig. 3 . Cell lysates were immunoprecipitated with an anti-p85 antibody followed by probing with anti-p85 and anti-IRS-1 antibodies in Western blot. Relative protein phosphorylation was analyzed by using Image J software. The results are the mean ± s.d. from three experiments. A77, A77 1726; * P < 0.05; ** P < 0.01.
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
- Download Figure
-
Download figure as PowerPoint slide
Immunoprecipitation
L6 myotubes and 3T3-L1 adipocytes were first starved of serum for 2 h or overnight, respectively, then incubated either in an amino-acid free medium (EBSS) or EBSS medium containing 2× amino acids (2×AA) in the absence or presence of A77 1726 for 5 h. Cells were left unstimulated or stimulated with insulin (100 nM) for 10 min. Cell lysates were immunoprecipitated with a rabbit monoclonal antibody against the p85 subunit of the PI-3 kinase, followed by probing with anti-p85 and anti-IRS1 antibodies in Western blot.
Confocal microscopy
Undifferentiated L6 cells seeded on coverslips were transiently transfected with mCherry-GLUT4-myc expression vector DNA using FuGENE6 following the manufacturer’s protocol. After incubation for 24 h, the cells were starved of serum for 2 h and then incubated in the absence or presence of A77 1726 (200 µM) for 4 h without or with 2× the amino acid concentrations in EBSS. Cells were left unstimulated or stimulated with 100 nM insulin for 30 min. The coverslips were collected, fixed and mounted with 50% glycerin in PBS containing 4,6-diamidino-2-phenylindole (DAPI) (0.5 µg/mL; Sigma Chemical). mCherry-tagged GLUT4 fluorescence was visualized under a Leica LP8 confocal microscope. The percent of cells positive for GLUT4 translocation into the plasma membrane among total mCherry-GLUT4-expressing cells was calculated by counting 10 randomly selected fields from each treatment. The results represent the mean ± standard deviation (s.d.) from one of three experiments with similar results.
Animals and drug administration
Use of animals and all experimental protocols were approved by the Institutional Animal Care and Use Committee of Rush University Medical Center and College of Veterinary Medicine, Yangzhou University. All mice were maintained on a 12-h light/darkness cycle and housed in ventilated cages at an ambient temperature of 23°C. Male ob/ob mice (B6.V-Lepob/OlaHsd, male) were purchased from Harlan Laboratories, Inc. These mice were fed ad libitum on a normal chow diet (NCD). CMC (1.5% dissolved in distilled water) was used as a vehicle to prepare leflunomide. Mice (8–10-week-old) were given 1.5% CMC or leflunomide by gavage. Uridine was co-administered with leflunomide to ob/ob mice to determine whether leflunomide could still control hyperglycemia when pyrimidine nucleotide levels in various tissues were normalized. The uridine dose was based on our previous studies (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib38 ">Xu et al. 1997, 1999 ) that 2 g/kg, twice daily is sufficient to normalize or overshoot pyrimidine nucleotide levels in the fast proliferating tumor cells or lymphocytes of lpr/lpr mice. Uridine dissolved in saline was given by intraperitoneal (i.p.) injection. ob/ob mice were treated with the vehicle (1.5% CMC daily), leflunomide (35 mg/kg/day, daily, gavage), uridine (2 g/kg, twice daily, i.p.) or leflunomide (35 mg/kg/day, daily, gavage) + uridine (2 g/kg, twice daily, i.p.) for three days. After fasting 6 h, blood was sampled from the tail vein and analyzed by a Bayer handheld glucometer. Body weight of mice was measured before and 3 days after treatment. Food intake was measured in the individual cages on day 3 after treatment.
For the glucose tolerance test (GTT) and insulin tolerance test (ITT), mice were treated for 3 days as above. On day 4, mice were fasted and treated with a last dose 6 h before GTT. Mice were challenged with glucose (1 g/kg) by intraperitoneal injection. For ITT, mice were similarly treated and fasted for 6 h, followed by intravenous injection of insulin (2.5 unit/kg) into the tail vein. Blood glucose levels were measured at various time points between 0 and 120 min (GTT) or between 0 and 60 min (ITT) by using a Bayer glucometer. AUC (area under curve) of the glucose levels in the GTT and ITT was calculated by using GraphPad Prism 5 software. AUC was plotted as bar graphs.
C57BL/6 male mice were purchased from the Center for Comparative Medicine, Yangzhou University. Mice (5 weeks old) were fed with NCD or HFD (24% fat, 24% protein, 41% carbohydrates, 1% others by weight, which translate to calories percentage as 20% protein, 35% carbohydrate, 45% fat) (Jiangsu Medicience Ltd, Yangzhou, China) for 10 weeks. Mice were treated with CMC or leflunomide followed by GTT as described earlier. ITT in C57BL/6 mice fed either NCD or HFD was conducted similarly as above except insulin (2.5 unit/kg) was intraperitoneally injected.
In vivo AKT activation
ob/ob mice were treated daily for three days. They were given a last dose on day 4 and then fasted for 6 h. Five minutes after insulin injection (2.5 unit/kg, intravenously), mice were sacrificed. Gastrocnemius muscle, mesenteric visceral white adipose and hepatic tissues (50–100 mg/sample) were collected and immediately homogenized in NP-40 lysis buffer. Protein concentrations were measured using a Pierce BCA Protein Assay kit (Thermo Fisher Scientific). AKTS473/T308, S6S235/236 and IRS-1S1101 phosphorylation was analyzed by Western blot. The density of the bands was analyzed by using NIH ImageJ software and normalized by the arbitrary units of their corresponding total proteins.
Statistical analysis
Data were presented as mean ± standard deviation (s.d.) (glucose uptake and GLUT4 translocation assays) or standard error of the mean (s.e.m.) (blood glucose levels). An unpaired Student t test was used to analyze the differences in glucose uptake in 3T3-L1 adipocytes and L6 myotubes in different groups, and the differences in the arbitrary number of Western blot data from the ImageJ analysis. Differences in blood glucose levels between different treatment groups were analyzed by repeated-measures ANOVA (analysis of variance). Differences in blood glucose levels in individual groups before and after treatment were statistically analyzed by a paired Student t test. A P value <0.05 was considered statistically significant. All statistics was performed with SigmaPlot 11 software (Systat Software, San Jose, CA, USA).
Results
A77 1726 induces feedback activation of the PI-3 kinase pathway
S6K1 inhibition by A77 1726 in tumor cell lines leads to feedback activation of the PI-3 kinase pathway through the insulin-like growth factor 1 (IGF-1) receptor (="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib11
">Doscas et al. 2014). Here, we tested if A77 1726 also induced feedback activation of the PI-3 kinase pathway in C2C12 myotubes. As shown in
Fig. 2
, insulin induced the phosphorylation of IRS-1S636, AKTT308, AKTS473 and S6K1T389 in C2C12 myotubes. A77 1726 enhanced insulin-induced phosphorylation of AKTS473, AKTT308 and S6K1T389, but inhibited the phosphorylation of S6S235/236 and IRSS1101, both of which are phosphorylated by S6K1. In contrast, A77 1726 increased the phosphorylation of IRSS636 (
Fig. 2
). This site is phosphorylated largely by mTOR. Of note, S6S235/236 and IRS-1S1101 were highly phosphorylated in unstimulated C2C12 myotubes. Rapamycin, an inhibitor of mTOR, increased AKTT308 and AKTS473 phosphorylation but inhibited IRSS1101, IRSS636, S6S235/236 and S6K1T389 phosphorylation. Increased S6K1 phosphorylation by A77 1726 is consistent with previous observations with PF-4708671, a specific S6K1 inhibitor (="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib23
">Pearce et al. 2010, ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib30
">Shum et al. 2016).
A77 1726 enhances insulin receptor signaling
We next examined the effect of A77 1726 on protein phosphorylation in the PI-3 kinase pathway in L6 myotubes and 3T3-L1 adipocytes in the presence of high amino acid concentrations, a condition of insulin resistance. As shown in
Fig. 3
, insulin induced AKTS473/T308 phosphorylation in the absence of amino acids in L6 myotubes. Consistent with previous observations (f="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib22
">Patti et al. 1998), high amino acids activated S6K1, as revealed by increased S6S235/236 and IRS-1S1101 phosphorylation, which is considered the indirect evidence of insulin resistance. A77 1726 induced phosphorylation of S6K1, which is a downstream molecule of mTOR, whereas rapamycin inhibited it. Both A77 1726 and rapamycin inhibited IRS-1S1101 and S6S235/236 phosphorylation but increased AKTS473 and AKTT308 phosphorylation in L6 myotubes. Similar results were obtained in 3T3-L1 adipocytes (
Fig. 4
).
href="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib34
">Um et al. (2004) reported that S6K1 deficiency leads to insulin receptor sensitization, as evidenced by increased insulin receptor tyrosine phosphorylation in the liver of insulin-treated mice, compared to that in wild-type mice. Here, we tested if A77 1726 could indeed enhance insulin receptor signaling. 3T3-L1 adipocytes and L6 myotubes preincubated with A77 1726 in the absence or presence of 2× amino acids and were left unstimulated or stimulated with insulin for 30 min. As shown in 5A, insulin-induced insulin receptor tyrosine phosphorylation in 3T3-L1 adipocytes and L6 myotubes that were cultured in the absence or presence of high concentrations of amino acids. This phosphorylation was further increased by A77 1726. Immunoprecipitation revealed that insulin stimulation increased the binding of the p85 subunit of the PI-3 kinase to IRS-1 in 3T3-L1 adipocytes and L6 myotubes cultured in the absence or presence of 2× amino acids. This binding was further increased by A77 1726 (
Fig. 5B
). Cell lysates were analyzed by Western blot. Equal levels of IRS-1 and the p85 subunit of the PI-3 kinase were found in the cell lysates used for immunoprecipitation (
Fig. 5B
).
A77 1726 increases GLUT4 translocation to the cell membrane
AKT activation induces the translocation of GLUT4 from the vesicles into the plasma membrane (ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib9
">Dann et al. 2007,
Copps & White 2012
). GLUT4 was detected in the cytoplasm of unstimulated L6 but was translocated to the plasma membrane in insulin-stimulated cells in the absence of amino acids (
Fig. 6
). A77 1726 alone slightly increased GLUT4 membrane translocation in L6 cells in the presence of 2× amino acids. Insulin also weakly induced GLUT4 translocation into the plasma membrane in the presence of 2× amino acids, almost as well as in the absence of 2× amino acids, probably due to the use of the undifferentiated L6 myoblast cells in this experiment. Alternatively, the relatively low transfection efficiency and a few GLUT4-positive cells under a high power field may shield the 2× amino acids-mediated inhibition of GLUT4 translocation. Nevertheless, A77 1726 significantly increased insulin-stimulated GLUT4 membrane translocation in L6 cells in the presence of 2× the amino acid concentrations (
Fig. 6
).
Figure 6
A77 1726 enhances insulin-induced GLUT4 translocation to the plasma membrane. (A) GLUT4 translocation to the plasma membrane. mCherry-GLUT4-myc-transfected L6 cells were starved of serum for 4 h and then incubated in the absence or presence of A77 1726 (200 µM) without or with 2× the amino acid concentrations. Cells were left unstimulated or stimulated with 20 nM insulin for 45 min. After fixation in methanol for 10 min, mCherry-tagged GLUT4 fluorescence was visualized under a Leica LP8 confocal microscope. Arrows denote the mCherry-tagged GLUT4 translocation to the cell membrane. (B) Quantification of the GLUT4 translocation to the plasma membrane. The data represent the mean ± s.d. from one of three experiments with similar results. * P < 0.05; ** P < 0.01. A full colour version of this figure is available at https:// doi.org/10.1530/JOE-17- 0536 .
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
- Download Figure
-
Download figure as PowerPoint slide
A77 1726 increases glucose uptake
We next determined if insulin receptor sensitization by A77 1726 led to increased glucose uptake. Cells treated with high concentrations of amino acids were considered under the insulin resistance condition. Indeed, 2× the amino acid concentrations significantly decreased the basal level of glucose uptake in L6 myotubes by 29% and reduced insulin-stimulated glucose uptake by 22%, compared to their corresponding controls ( Fig. 7A ). A77 1726 increased insulin-stimulated glucose uptake in the presence of 2× the amino acid concentrations by 31%. Rapamycin included as a positive control increased glucose uptake by 19% ( Fig. 7A ) in insulin-stimulated L6 myotubes in the presence of 2× the amino acid concentrations. Slightly better stimulation of insulin-induced glucose uptake by A77 1726 was observed with 3T3-L1 adipocytes ( Fig. 7B ). Of note, A77 1726 is a cytostatic drug and does not affect the viability and proliferation of differentiated non-dividing cells. Cells were incubated in the presence of A77 1726 only for a few hours. Using an Enhanced Cell Counting Kit-8 (CCK-8), we found that A77 1726 (200 µM) did not have any cytotoxicity on 3T3-L1 adipocytes and L6 myotubes (data not shown). Using a CellTiter-Glo Luminescent Cell Viability Assay, we found that A77 1726 (200 µM) reduced the proliferation of 3T3-L1 adipocytes by approximately 20% after incubation for 48 h (data not shown).
Figure 7
A77 1726 increases glucose uptake. L6 myotubes (A) and 3T3-L1 adipocytes (B) seeded in 24-well plates were starved in serum-free, low glucose DMEM medium for 4 h. The cells were then incubated in EBSS in the absence or presence of 2×AA and/or A77 1726 (200 µM) or rapamycin (50 nM) for 2 h. The cells were left unstimulated or stimulated with 20 nM insulin for 45 min followed by incubation of [3H]-2-DG for 5 min. The data are the mean ± s.d. of the triplicate in one of three experiments with similar results. * P < 0.05; ** P < 0.01, compared to the control (with insulin and 2× amino acids but without drug).
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
- Download Figure
-
Download figure as PowerPoint slide
Control of hyperglycemia by leflunomide
We first assessed the ability of leflunomide to control hyperglycemia in male mice since estrogen in female mice may protect against high-fat diet-induced metabolic syndrome. Female animals are prone to adipose tissue storage and glucose homeostasis, whereas males are predisposed to diabetes. As shown in Table 2 , blood glucose levels were very high (>200 mg/dL) in ob/ob mice before treatment and remained very high in mice treated with CMC, a vehicle used to dissolve leflunomide. Fasting blood glucose levels decreased to normal levels in mice treated with leflunomide alone or with leflunomide plus uridine (<110 mg/dL). Statistical analysis revealed that leflunomide treatment significantly lowered blood glucose levels ( P < 0.01). Treatment with uridine, a nucleoside used to normalize pyrimidine nucleotide levels in vitro and in vivo (href=" https:// joe.bioscientifica.com/ view/journals/joe/237/1/JOE-17-0536.xml#bib38 ">Xu et al. 1997, 1999 ), had no effect on fasting blood glucose levels and did not block leflunomide-mediated control of hyperglycemia. These observations suggest that leflunomide controls hyperglycemia independent of its inhibitory effect on pyrimidine nucleotide synthesis. There were no significant differences in food intake and body weight among mice treated with CMC, leflunomide, uridine or leflunomide plus uridine for 3 days ( Table 2 ).
Table 2Control of hyperglycemia by leflunomide in ob/ob mice.†
| Drug | Number of mice (N) | Blood glucose concentration (mg/dL) | Body weight (g) | Food intake (g/day) | P value* | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Before | After | P value | Before | After | Change | P value* | ||||
| Vehicle | 6 | 234 ± 16 | 252 ± 22 | 0.697 | 47.8 ± 2.9 | 48.0 ± 3.1 | 0.12 ± 0.21 | 4.9 ± 0.23 | ||
| Leflunomide | 7 | 275 ± 12 | 111 ± 6 | 0.001 | 48.4 ± 0.48 | 48.5 ± 0.51 | −0.02 ± 0.13 | 0.252 | 5.0 ± 0.09 | 0.687 |
| Uridine | 4 | 210 ± 12 | 253 ± 15 | 0.153 | 48.7 ± 0.68 | 48.5 ± 0.69 | −0.02 ± 0.08 | 0.793 | 4.9 ± 0.07 | 1.000 |
| Lef + Uridine | 4 | 222 ± 15 | 97 ± 15 | 0.021 | 48.5 ± 1.21 | 48.4 ± 1.06 | −0.3 ± 0.04 | 0.385 | 4.8 ± 0.04 | 0.272 |
†
ob/ob
mice (male, 8–10 weeks) were treated with the vehicle (1.5% CMC, daily, gavage), leflunomide (35 mg/kg, daily, gavage), uridine (2 g/kg, twice daily, i.p.) or leflunomide (35 mg/kg, daily, gavage) + uridine (2 g/kg, twice daily, i.p.) for three days. After fasting for 6 h, blood glucose levels were measured and statistically analyzed by a paired Student
t
test. Body weight of mice were measured before and 3 days after treatment. Food consumption was measured on individual mouse in a separate cage on day 3 after treatment.
*
Compared to the vehicle control.
We next conducted GTT to examine the ability of leflunomide to lower blood glucose levels in
ob/ob
mice. Blood glucose levels in mice receiving leflunomide or leflunomide plus uridine were significantly lower than those treated with CMC (
P
< 0.001) (
Fig. 8A
). Uridine treatment alone did not significantly alter blood glucose levels in mice. ITT assay revealed that blood glucose levels were elevated in control mice or in mice treated with uridine 15 min after insulin injection (
Fig. 8C
). In contrast, insulin did not increase blood glucose levels in leflunomide-treated mice but was able to slightly increase blood glucose levels in mice treated with uridine plus leflunomide (
Fig. 8C
). Transient increase of blood glucose levels after insulin injection into the tails of control
ob/ob
mice is probably caused by stress or due to the insulin receptor internalization. Transient increase of blood glucose levels after insulin injection is consistent with observations made by others (href="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib17
">He et al. 2013). AUC was significantly decreased in leflunomide- or leflunomide plus uridine-treated mice, compared to those treated with CMC or uridine alone (
Fig. 8B
and
D
) (
P
< 0.01). Consistently, leflunomide treatment significantly decreased blood glucose levels in GTT (
Fig. 8E
and
F
) and ITT assays (
Fig. 8G
and
H
) in HFD-fed mice, compared to those treated with CMC. Leflunomide appears more effective in lowering blood glucose levels in
ob/ob
mice while only modestly lowering blood glucose levels in the mice with HFD-induced diabetes. Leflunomide treatment did not significantly alter blood glucose levels in mice fed a NCD (
Fig. 8E
,
F
,
G
and
H
).
Figure 8
Control of hyperglycemia and insulin receptor sensitization by leflunomide. (A) ob/ob mice were treated as described in ‘Materials and methods’ section. The mice were challenged with glucose (1 g/kg) by intraperitoneal injection for GTT. Blood glucose levels were measured at the indicated time. CMC vs leflunomide, uridine or leflunomide + uridine, P < 0.001; leflunomide vs leflunomide + uridine, P = 0.002. Lef, leflunomide; Uri, uridine; The arbitrary values of AUC were calculated and shown in a bar graph (B) **CMC vs leflunomide or leflunomide + uridine, P < 0.001; CMC vs uridine, P = 0.057. (C) Control of hyperglycemia by leflunomide in ITT. ob/ob mice were treated as described in ‘Materials and methods’ section. After fasting for 6 h, insulin (2.5 U/kg) was injected intravenously. Blood glucose levels were measured at the indicated times. CMC vs leflunomide, uridine or leflunomide + uridine, P < 0.001; leflunomide vs leflunomide + uridine, P = 0.006. (D) AUC in the different treatment groups was shown as the bar graph. **CMC vs leflunomide or leflunomide + uridine, P < 0.001; CMC vs uridine, P = 0.115. (E and G) C57BL/6 male mice (5w-weeks-old) fed on normal chow diet (NCD) or high-fat diet (HFD) for 10 weeks were treated daily with CMC or leflunomide (35 mg/kg/day) for 3 days and then evaluated for blood glucose levels in GTT (E) and ITT (G). (F and H) AUC in different treatment group was shown as bar graphs. **CMC vs leflunomide in mice fed with HFD, P < 0.001; CMC vs leflunomide in mice fed with NCD, P > 0.05. A full colour version of this figure is available at https:// doi.org/10.1530/JOE-17- 0536 .
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
- Download Figure
-
Download figure as PowerPoint slide
Finally, we tested if leflunomide treatment enhanced insulin receptor signaling in vivo in ob/ob mice. As shown in Fig. 9A , insulin significantly increased AKTS473/T308 phosphorylation in the muscle, fat and liver of normal mice, compared to the unstimulated controls ( P < 0.05) ( Fig. 9C ). Leflunomide treatment did not or only slightly enhanced insulin-induced AKT phosphorylation in these insulin-sensitive tissues of control mice. The levels of AKTS473/T308 phosphorylation in the tissue of the control and leflunomide-treated mice were comparable ( Fig. 9A and C ) ( P > 0.05). In contrast, insulin poorly induced AKTS473/T308 phosphorylation in the muscle, fat and liver of ob/ob mice ( Fig. 9B ). Densitometry analysis revealed that AKTS473/T308 phosphorylation in the insulin-sensitive tissue was not significantly different between the untreated and insulin-treated mice ( P > 0.05) ( Fig. 9D ). Leflunomide treatment significantly increased insulin-stimulated AKTS473/T308 phosphorylation in the muscle and adipose tissues, compared to those mice treated with the control vehicle only ( P < 0.05). AKTS473/T308 phosphorylation levels appeared to be higher in the liver of leflunomide-treated ob/ob mice than that of untreated ob/ob mice. However, statistical analysis did not reach significance. Similarly, AKTS473/T308 phosphorylation levels were also higher in the muscle, fat and liver of ob/ob mice treated with leflunomide plus uridine than those treated with uridine alone (Supplementary Fig. 1, see section on supplementary data given at the end of this article). However, there was no significant difference in S6 and IRS-1 phosphorylation in the muscle, fat and liver between untreated and leflunomide-treated mice (Supplementary Fig. 2).
Figure 9
AKT phosphorylation in the metabolic tissues is enhanced by leflunomide. C57BL/6 (A) and ob/ob (B) mice were treated daily with CMC (1.5%) or leflunomide (35 mg/kg/day) for 3 days. On day 4, the mice were treated with a last dose and then fasted for 6 h. Mice were then injected intravenously with saline or insulin (2.5 unit/kg). Five minutes later, the mice were sacrificed. Gastrocnemius muscle, visceral white adipose and liver tissues were harvested and analyzed for AKTS473/T308 phosphorylation by Western blot with their specific antibodies and reprobed with an antibody against total AKT protein. Immunoblots represent the data from two animals in each group. The experiment was repeated twice. (C and D) The density of the bands in the tissues of normal C57BL/6 mice (C) and ob/ob mice (D) was analyzed by using NIH Image-J software and normalized by the arbitrary units of their corresponding total proteins. The results are the mean ± s.d. from four animals in each group. * P < 0.05; ns, not significant between CMC/insulin-treated and leflunomide/insulin-treated group.
Citation: Journal of Endocrinology 237, 1; 10.1530/JOE-17-0536
Discussion
S6K1 hyperactivation has been implicated in insulin resistance and the development of hyperglycemia and obesity (href="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib34
">Um et al. 2004,
2006
). S6K1-deficient mice are resistant to HFD-induced hyperglycemia and obesity (href="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib34
">Um et al. 2004,
2006
). ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib30
">Shum et al. (2016) recently reported that PF-4708671, an S6K1 inhibitor, controls hyperglycemia in HFD-fed mice by sensitizing the insulin receptor. We recently identified S6K1 as a molecular target of leflunomide and A77 1726 (="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib11
">Doscas et al. 2014). Inhibition of S6K1 activity by A77 1726 in A375 melanoma cells leads to the feedback activation of the PI-3 kinase pathway, as evidenced by increased AKTS473 and decreased S6S235/236 phosphorylation. Our present study showed that inhibition of S6K1 activity by A77 1726 led to the inhibition of S6S235/236 and IRS-1S1101 phosphorylation in mouse C2C12 and rat L6 myotubes and in mouse 3T3-L1 adipocytes under normal and/or insulin resistance conditions. A77 1726 enhanced insulin-induced insulin receptor tyrosine phosphorylation, binding of the p85 subunit of the PI-3 kinase to IRS-1 (
Fig. 5
), and insulin-induced GLUT4 translocation to the plasma membrane in L6 cells in the presence of high amino acid concentrations (
Fig. 6
). Leflunomide treatment increased AKTT308/S473 phosphorylation in the muscular and adipose tissues
in vivo
(Fig. 9B)
. These observations collectively suggest that inhibition of S6K1 by A77 1726 is primarily responsible for its anti-hyperglycemic effect. ref="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib16
">Guo et al. (1997) reported earlier that chronic use of leflunomide has no effect on blood glucose levels in healthy rats. Because there is no insulin resistance in the insulin-sensitive tissues of normal animals, leflunomide cannot further sensitize the insulin receptor. Consistently, we found that there was no significant difference in blood glucose levels in NCD-fed lean mice treated with CMC or leflunomide (
Fig. 8E
,
F
,
G
and
H
).
We investigated the inhibitory effect of A77 1726 on the phosphorylation of two relevant substrates of S6K1, S6S235/236 and IRS-1S1101. Due to feedback activation of the PI-3 kinase pathway, S6K1 phosphorylation was increased in A77 1726-treated cells. A77 1726 inhibited S6S235/236 and IRS-1S1101 phosphorylation in myotubes and adipocytes
in vitro
. In contrast, leflunomide treatment did not significantly decrease S6S235/236 and IRS-1S1101 in the metabolic tissues of insulin-stimulated mice
in vivo
(Supplementary Fig. 2). Similar to our observations, (ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib30
">Shum et al. 2016) reported that PF-4708671, a more potent inhibitor of S6K1 than A77 1726, did not inhibit S6 phosphorylation
in vivo
. These investigators suggest that compensatory S6K2 activation may shield the inhibitory effect of PF-4708671 on S6 phosphorylation (ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib30
">Shum et al. 2016). ="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib23
">Pearce et al. (2010) reported that PF-4708671 did not inhibit early S6 phosphorylation in 293 cells in the presence of serum. We speculate that inhibition of S6K1 activity by A77 1726 or PF-4708671 may initially inhibit S6 and IRS-1 phosphorylation. However, when S6K1 is further feedback activated, incomplete inhibition of S6 activity may account for lack of inhibition of S6 and IRS-1 phosphorylation. Within a few minutes after insulin injection, leaked S6K1 activity is sufficient to fully phosphorylate S6 and IRS-1. However, once AKT is feedback activated, AKT phosphorylation levels will remain high for a while. Activated AKT continues to regulate glucose metabolism until it is dephosphorylated and inactivated. IRS-1 is phosphorylated at S636 by mTOR (
Copps & White 2012
). Our present study showed that due to feedback activation of mTOR, A77 1726 induced IRS-1S636 phosphorylation in C2C12 myotubes. Interestingly, IRS-1S636 phosphorylation by ROCK1 leads to the activation of PI-3 kinase (
Copps & White 2012
). Thus, A77 1726 may improve insulin receptor signaling by inhibiting IRS-1S1101 and by increasing IRS-1S636 phosphorylation.
Leflunomide and A77 1726 inhibit S6K1 activity with the IC50 values of approximately 50–75 µM (="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib11
">Doscas et al. 2014). Plasma concentrations of A77 1726 in RA patients treated with leflunomide (20 mg/day) are higher than 200 µM (ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib5
">Chan et al. 2005). A77 1726 in the blood of mice treated with leflunomide at a dose of 35 mg/kg had a remarkably long half-life of 15 h. The blood concentrations of A77 1726 reached a peak of 500 µM within 4 h and remained at 250 µM at 24 h after a single dose of 35 mg/kg of leflunomide in mice (f="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib6
">Chong et al. 1999). These data suggest that A77 1726 concentrations in plasma are high enough to inhibit S6K1 activity. Indeed, AKT phosphorylation was increased in the metabolic tissues of leflunomide-treated mice. Blood glucose levels were decreased to normal levels in
ob/ob
mice treated with leflunomide at a dose of 35 mg/kg/day. Co-administration with uridine was unable to block the anti-glycemic effect of leflunomide. An earlier clinical study revealed that the mean glucose levels in the blood of RA patients treated with leflunomide (83 mg/dL) were significantly lower than those treated with other regimens (93 mg/dL) (ref="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib25
">Rho et al. 2009). About 10% of patients treated with leflunomide undergo weight loss that cannot be attributed to diarrhea or other gastrointestinal side effects and is likely due to an increased metabolic rate (="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib7
">Coblyn et al. 2001). These observations collectively suggest that A77 1726 concentrations are high enough to inhibit S6K1 activity, subsequently leading to better control of hyperglycemia. Inflammatory cytokines, in particular TNF-α, can also desensitize the insulin receptor and contribute to the development of hyperglycemia (
Copps & White 2012
). Anti-TNF-α therapy does not lower blood glucose levels in RA patients (
Rosenvinge et al. 2007
). Many anti-inflammatory drugs, unlike leflunomide, do not lower blood glucose levels (ref="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib25
">Rho et al. 2009), suggesting that inhibition of inflammation is not a sufficient explanation for better control of hyperglycemia.
We are aware of several weaknesses in our study. First, the evidence supporting the conclusion that leflunomide sensitized the insulin receptor and controlled hyperglycemia by inhibiting the activity of S6K1
in vivo
is weak. Increased AKT phosphorylation in the tissue of leflunomide-treated mice with insulin resistance indirectly suggests feedback activation of the PI-3 kinase pathway due to the inhibition of S6K1 activity. However, inhibition of S6 and IRS-1 phosphorylation was not seen in the tissue of leflunomide-treated animals. This is consistent with the observations in a study using PF-4708671, a potent S6K1 inhibitor (ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib30
">Shum et al. 2016). Second, leflunomide worked extremely well in lowering blood glucose levels in the
ob/ob
mice, even with a relatively small sample size, the
P
values reached statistically significant levels. However, leflunomide only modestly decreased blood glucose levels in the GTT and ITT in the mouse model of the HFD-induced diabetes. Third, leflunomide treatment significantly enhanced insulin-stimulated AKT phosphorylation in the muscle and adipose tissues of
ob/ob
mice. Leflunomide treatment increased AKT phosphorylation in the liver of
ob/ob
mice, but this was not statistically significant due to a relatively small number of animals and the high variability of AKT phosphorylation. Fourth, since there are already many anti-diabetic drugs available for treating hyperglycemia, leflunomide might be only useful for a certain type of patient population such as those with both diabetes and RA. The diabetes mouse models used in our study do not have RA, thus not completely recapitulating a clinical setting in which leflunomide deems to be used.
Chronic use of rapamycin does not control but rather exacerbates hyperglycemia in mouse models and in patients (
Di Paolo et al. 2006
,
Schindler et al. 2014
). Failure of rapamycin to control hyperglycemia is, after chronic use, due to its ability to inhibit the activity of mTORC2 (
Fig. 1B
), which is responsible for phosphorylating AKTS473 (
"https://joe.bioscientifica.com/view/journals/joe/237/1/JOE-17-0536.xml#bib20">Lamming et al. 2012
). In the present study, we did not follow-up the anti-hyperglycemic effect after chronic use of leflunomide. However, several lines of evidence suggest that chronic use of leflunomide will likely maintain its therapeutic efficacy: (1) Unlike rapamycin, A77 1726 inhibits S6K1 (="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib11
">Doscas et al. 2014), a kinase downstream of mTOR. Therefore, A77 1726 does not act like rapamycin to inhibit mTORC2 (
Fig. 1B
); (2) The anti-hyperglycemic effect of PF-4708671 after chronic use is sustainable
in vivo
(ef="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib30
">Shum et al. 2016). Both A77 1726 and PF-4708671 control hyperglycemia by inhibiting S6K1; (3) Blood glucose levels are significantly lower in RA patients chronically treated with leflunomide than those treated with other drugs (ref="
https://
joe.bioscientifica.com/
view/journals/joe/237/1/JOE-17-0536.xml#bib25
">Rho et al. 2009). Nevertheless, whether leflunomide can maintain a long-lasting therapeutic effect after chronic use should be verified first in animal models before it can be investigated in patients.
In summary, our present study showed that A77 1726 induced feedback activation of the PI-3 kinase pathway by inhibiting S6K1 activity and that A77 1726 increased glucose uptake and GLUT4 translocation to the plasma membrane
in vitro
(
Fig. 1
B). A77 1726 may also improve glucose metabolisms by stimulating glycogen synthesis and by inhibiting gluconeogenesis through activated AKT (
Fig. 1B
). Leflunomide was capable of controlling hyperglycemia and improving insulin sensitivity
in vivo
. Leflunomide could be particularly useful for treating RA patients who also have type 2 diabetes.
Supplementary data
This is linked to the online version of the paper at
https://
doi.org/10.1530/JOE-17-
0536
.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by grants from Rush Pilot Diabetes Research fund, Natural Science Foundation of China (81672463) and the Priority Academic Program Development of Jiangsu Higher Education Institutions to X Xu, and by an NIH R01 (CA204926) grant to Y Li.
Author contribution statement
X X conceived concept, wrote manuscript and researched data. J C; J S, M E D, J Y; A J W conducted experiments and contributed to discussion; Y L (University of Chicago) contributed to discussion; Y L (Baylor College of Medicine) R A P contributed to discussion, reviewed and edited the manuscript.
Acknowledgements
The authors are very grateful to Dr James W. Williams in CinKate Corporation for kindly providing A77 1726 and leflunomide and Dr Amira Klip (The Hospital for Sick Children, Toronto, Ontario) for mCherry-GLUT4-myc expression vector.
References
-
Boura-Halfon S & Zick Y 2009 Phosphorylation of IRS proteins, insulin action, and insulin resistance.
American Journal of Physiology-Endocrinology and Metabolism
296 E581–E591. (
https://
doi.org/10.1152/ajpendo
.90437.2008
)
-
Breedveld FC & Dayer JM 2000 Leflunomide: mode of action in the treatment of rheumatoid arthritis.
Annals of the Rheumatic Diseases
59 841–849. (
https://
doi.org/10.1136/ard.59.
11.841
)
-
Bruneau JM, Yea CM, Spinella-Jaegle S, Fudali C, Woodward K, Robson PA, Sautes C, Westwood R, Kuo EA, Williamson RA, et al.1998 Purification of human dihydro-orotate dehydrogenase and its inhibition by A77 1726, the active metabolite of leflunomide.
Biochemical Journal
336 299–303. (
https://
doi.org/10.1042/bj33602
99
)
-
Cannon GW & Kremer JM 2004 Leflunomide.
Rheumatic Diseases Clinics of North America
30 295–309. (
https://
doi.org/10.1016/j.rdc.2
004.01.010
)
-
Chan V, Charles BG & Tett SE 2005 Population pharmacokinetics and association between A77 1726 plasma concentrations and disease activity measures following administration of leflunomide to people with rheumatoid arthritis.
British Journal of Clinical Pharmacology
60 257–264. (
https://
doi.org/10.1111/j.1365-
2125.2005.02415.x
)
-
Chong AS, Huang W, Liu W, Luo J, Shen J, Xu W, Ma L, Blinder L, Xiao F, Xu X, et al.1999 In vivo activity of leflunomide: pharmacokinetic analyses and mechanism of immunosuppression.
Transplantation
68 100–109. (
https://
doi.org/10.1097/0000789
0-199907150-00020
)
-
Coblyn JS, Shadick N & Helfgott S 2001 Leflunomide-associated weight loss in rheumatoid arthritis.
Arthritis and Rheumatology
44 1048–1051. (
https://
doi.org/10.1002/1529-01
31(200105)
44:5<1048::AID-ANR184>3.0.CO;2-V)
-
Copps KD & White MF 2012 Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2.
Diabetologia
55 2565–2582. (
https://
doi.org/10.1007/s00125-
012-2644-8
)
-
Dann SG, Selvaraj A & Thomas G 2007 mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer.
Trends in Molecular Medicine
13 252–259. (
https://
doi.org/10.1016/j.molme
d.2007.04.002
)
-
Di Paolo S, Teutonico A, Leogrande D, Capobianco C & Schena PF 2006 Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: a crossroad between cancer and diabetes?
Journal of the American Society of Nephrology
17 2236–2244. (
https://
doi.org/10.1681/ASN.200
6030196
)
-
Doscas ME, Williamson AJ, Usha L, Bogachkov Y, Rao GS, Xiao F, Wang Y, Ruby C, Kaufman H, Zhou J, et al.2014 Inhibition of p70 S6 kinase (S6K1) activity by A77 1726 and its effect on cell proliferation and cell cycle progress.
Neoplasia
16 824–834. (
https://
doi.org/10.1016/j.neo.2
014.08.006
)
-
Elder RT, Xu X, Williams JW, Gong H, Finnegan A & Chong AS 1997 The immunosuppressive metabolite of leflunomide, A77 1726, affects murine T cells through two biochemical mechanisms.
Journal of Immunology
159 22–27.
-
Fenton TR & Gout IT 2010 Functions and regulation of the 70kDa ribosomal S6 kinases.
International Journal of Biochemistry and Cell Biology
43 47–59. (
https://
doi.org/10.1016/j.bioce
l.2010.09.018
)
-
Guo S 2013 Molecular basis of insulin resistance: the role of IRS and Foxo1 in the control of diabetes mellitus and its complications.
Drug Discovery Today: Disease Mechanisms
10 e27–e33. (
https://
doi.org/10.1016/j.ddmec
.2013.06.003
)
-
Guo S 2014 Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms.
Journal of Endocrinology
220 T1–T23. (
https://
doi.org/10.1530/JOE-13-
0327
)
-
Guo Z, Chong AS, Shen J, Foster P, Sankary HN, McChesney L, Mital D, Jensik SC, Gebel H & Williams JW 1997 In vivo effects of leflunomide on normal pancreatic islet and syngeneic islet graft function.
Transplantation
63 716–721. (
https://
doi.org/10.1097/0000789
0-199703150-00018
)
-
He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O’Doherty RM & Xie W 2013 PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice.
Diabetes
62 1876–1887. (
https://
doi.org/10.2337/db12-10
39
)
-
Herlitz-Cifuentes HS, Garces PC, Fernandez LI & Guzman-Gutierrez EA 2015 Effect of systemic inflammation on the function of insulin and glucose metabolism in rheumatoid arthritis.
Current Diabetes Reviews
12 156–162. (
https://
doi.org/10.2174/1573399
811666150602150325
)
-
Jiang P, Li H & Li X 2015 Diabetes mellitus risk factors in rheumatoid arthritis: a systematic review and meta-analysis.
Clinical and Experimental Rheumatology
33 115–121.
-
Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, et al.2012 Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity.
Science
335 1638–1643. (
https://
doi.org/10.1126/science
.1215135
)
-
Nathan DM 2015 Diabetes: advances in diagnosis and treatment.
JAMA
314 1052–1062. (
https://
doi.org/10.1001/jama.20
15.9536
)
-
Patti ME, Brambilla E, Luzi L, Landaker EJ & Kahn CR 1998 Bidirectional modulation of insulin action by amino acids.
Journal of Clinical Investigation
101 1519–1529. (
https://
doi.org/10.1172/JCI1326
)
-
Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C & Alessi DR 2010 Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1).
Biochemical Journal
431 245–255. (
https://
doi.org/10.1042/BJ20101
024
)
-
Pinto AJ, Roschel H, de Sa Pinto AL, Lima FR, Pereira RMR, Silva CA, Bonfa E & Gualano B 2017 Physical inactivity and sedentary behavior: overlooked risk factors in autoimmune rheumatic diseases?
Autoimmunity Reviews
16 667–674. (
https://
doi.org/10.1016/j.autre
v.2017.05.001
)
-
Rho YH, Oeser A, Chung CP, Milne GL & Stein CM 2009 Drugs used in the treatment of rheumatoid arthritis: relationship between current use and cardiovascular risk factors.
Archives of Drug Information
2 34–40. (
https://
doi.org/10.1111/j.1753-
5174.2009.00019.x
)
-
Rosenvinge A, Krogh-Madsen R, Baslund B & Pedersen BK 2007 Insulin resistance in patients with rheumatoid arthritis: effect of anti-TNFalpha therapy.
Scandinavian Journal of Rheumatology
36 91–96. (
https://
doi.org/10.1080/0300974
0601179605
)
-
Ruckemann K, Fairbanks LD, Carrey EA, Hawrylowicz CM, Richards DF, Kirschbaum B & Simmonds HA 1998 Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans.
Journal of Biological Chemistry
273 21682–21691. (
https://
doi.org/10.1074/jbc.273
.34.21682
)
-
Schindler CE, Partap U, Patchen BK & Swoap SJ 2014 Chronic rapamycin treatment causes diabetes in male mice.
American Journal of Physiology: Regulatory, Integrative and Comparative Physiology
307 R434–R443. (
https://
doi.org/10.1152/ajpregu
.00123.2014
)
-
Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, et al.2009 Ribosomal protein S6 kinase 1 signaling regulates mammalian life span.
Science
326 140–144. (
https://
doi.org/10.1126/science
.1177221
)
-
Shum M, Bellmann K, St-Pierre P & Marette A 2016 Pharmacological inhibition of S6K1 increases glucose metabolism and Akt signalling in vitro and in diet-induced obese mice.
Diabetologia
59 592–603. (
https://
doi.org/10.1007/s00125-
015-3839-6
)
-
Siemasko KF, Chong AS, Williams JW, Bremer EG & Finnegan A 1996 Regulation of B cell function by the immunosuppressive agent leflunomide.
Transplantation
61 635–642. (
https://
doi.org/10.1097/0000789
0-199602270-00020
)
-
Siemasko K, Chong AS, Jack HM, Gong H, Williams JW & Finnegan A 1998 Inhibition of JAK3 and STAT6 tyrosine phosphorylation by the immunosuppressive drug leflunomide leads to a block in IgG1 production.
Journal of Immunology
160 1581–1588.
-
Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al.2004 Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity.
Nature
431 200–205. (
https://
doi.org/10.1038/nature0
2866
)
-
Um SH, D’Alessio D & Thomas G 2006 Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1.
Cell Metabolism
3 393–402. (
https://
doi.org/10.1016/j.cmet.
2006.05.003
)
-
Versini M, Jeandel PY, Rosenthal E & Shoenfeld Y 2014 Obesity in autoimmune diseases: not a passive bystander.
Autoimmunity Reviews
13 981–1000. (
https://
doi.org/10.1016/j.autre
v.2014.07.001
)
-
Williamson RA, Yea CM, Robson PA, Curnock AP, Gadher S, Hambleton AB, Woodward K, Bruneau JM, Hambleton P, Spinella-Jaegle S, et al.1996 Dihydroorotate dehydrogenase is a target for the biological effects of leflunomide.
Transplantation Proceedings
28 3088–3091.
-
Xia T, Cheng Y, Zhang Q, Xiao F, Liu B, Chen S & Guo F 2012 S6K1 in the central nervous system regulates energy expenditure via MC4R/CRH pathways in response to deprivation of an essential amino acid.
Diabetes
61 2461–2471. (
https://
doi.org/10.2337/db11-12
78
)
-
Xu X, Williams JW, Bremer EG, Finnegan A & Chong AS 1995 Inhibition of protein tyrosine phosphorylation in T cells by a novel immunosuppressive agent, leflunomide.
Journal of Biological Chemistry
270 12398–12403. (
https://
doi.org/10.1074/jbc.270
.21.12398
)
-
Xu X, Williams JW, Gong H, Finnegan A & Chong AS 1996 Two activities of the immunosuppressive metabolite of leflunomide, A77 1726. Inhibition of pyrimidine nucleotide synthesis and protein tyrosine phosphorylation.
Biochemical Pharmacology
52 527–534. (
https://
doi.org/10.1016/0006-29
52(96)00303-6
)
-
Xu X, Blinder L, Shen J, Gong H, Finnegan A, Williams JW & Chong AS 1997 In vivo mechanism by which leflunomide controls lymphoproliferative and autoimmune disease in MRL/MpJ-lpr/lpr mice.
Journal of Immunology
159 167–174.
-
Xu X, Shen J, Mall JW, Myers JA, Huang W, Blinder L, Saclarides TJ, Williams JW & Chong AS 1999 In vitro and in vivo antitumor activity of a novel immunomodulatory drug, leflunomide: mechanisms of action.
Biochemical Pharmacology
58 1405–1413. (
https://
doi.org/10.1016/S0006-2
952(99)00228-2
)
-
Zebisch K, Voigt V, Wabitsch M & Brandsch M 2012 Protocol for effective differentiation of 3T3-L1 cells to adipocytes.
Analytical Biochemistry
425 88–90. (
https://
doi.org/10.1016/j.ab.20
12.03.005
)
-
Zimmet P, Alberti KG & Shaw J 2001 Global and societal implications of the diabetes epidemic.
Nature
414 782–787. (
https://
doi.org/10.1038/414782a
)
-
Zimmet PZ, Magliano DJ, Herman WH & Shaw JE 2014 Diabetes: a 21st century challenge.
Lancet Diabetes and Endocrinology
2 56–64. (
https://
doi.org/10.1016/S2213-8
587(13)70112-8
)
谷歌镜像检索 A771726 site:joe.bioscientifica.com 得到该文献 [11]
扬州大学教师个人主页服务平台 徐秀龙--中文主页--专利 (yzu.edu.cn) [12]
徐秀龙 , A771726(特立氟胺 [13] )在制备治疗流感病毒感染相关疾病的药物中的应用
徐秀龙 , 来氟米特或其活性代谢产物A771726(特立氟胺 [13] )在制备治疗2型糖尿病的药物中的应用
类风湿消炎药也可降糖-疾病防控 榆林市卫生健康委员会 索引号:013294 来源:生命时报 2018-03-23(yl.gov.cn) [14] [15]
慢性自身免疫性疾病类风湿性关节炎患者约占全世界人口的1%,该病可导致关节疼痛和肿胀。来氟米特(Arava) [16] 是治疗类风湿关节炎的一种常见消炎药。美国《内分泌学杂志》近日刊登我国一项新研究称,风湿性关节炎常见药物来氟米特还有助于降低血糖水平。
新研究由中国扬州大学比较医学研究所 徐秀龙 [17] {徐休龙(音译)} [18] 教授及其研究小组完成。实验鼠模型研究发现,来氟米特可使患有2型糖尿病的实验鼠血糖水平降低,胰岛素抵抗逆转,原因是其可应对导致胰岛素受体不敏感的一种蛋白质,而胰岛素受体的作用正是指导细胞吸收血液中的糖分。这一实验结果表明,来氟米特可作为一种有效的降糖药物,该疗法特别适合同时患糖尿病和类风湿性关节炎的患者。
徐教授表示,来氟米特早已被批准用于治疗类风湿性关节炎,多项早期临床研究也发现,服用该药的肥胖患者可在一定程度上有效减肥。更多深入研究尚待完成,徐教授团队下一步将继续展开实验鼠研究以及人体临床试验。 (金也)

尝试找所给的出处链接替代链接

如何看待扬州大学关于来氟米特在糖尿病治疗上的研究? [20] @罕见病友临行疾呼

site:sina.cn 扬州大学 来氟米特 糖尿病_百度搜索 [21]
5种常见的疾病根本无法治愈,说能治愈的或只想骗钱,别上当受骗|强直性脊柱炎|多囊卵巢综合征|牛皮癣_新浪新闻 (sina.com.cn) [22]
"扬州大学" "来氟米特" "糖尿病"_百度搜索 [23]





"扬州大学" "来氟米特" "糖尿病"中国政府网百度检索结果 site:gov.cn [24]

"扬州大学" "来氟米特" "糖尿病" site:edu.cn_百度搜索 [25]
ref=" http://www. baidu.com/link? url=s0_5Pvrwy8ueSPYeMMozmISRvktjNiiDNQHjyfXVJErEzZKwsZI6FzY1W2e8nalUM9bbjMMxHGr7pp7LBdwSnKmE_HRZmL-DPKOzTYH61yq ">扬州大学教师个人主页服务平台 徐秀龙--中文主页-- 来氟米...
2020年12月9日 徐秀龙,XXL, 扬州大学 教师个人主页服务平台, 来氟米特 或其活性代谢产物A771726在制备治疗2型 糖尿病 的药物中的应用徐秀龙, teacher.yzu.edu.cn/XXL1/zh_CN/...
ref=" http://www. baidu.com/link? url=Aw-CIoYk8n0lRVOy4Yl_Jq9zUUrHJREhWAfEEnUGSYflW5hSMd9RHp1HrbTl0YHrXnsgXIftD7-fm3uEkctdchO859unLj9n5tpDI8j6v-7 ">扬州大学教师个人主页服务平台 XXL--Home-- A771726在制备...
徐秀龙,XXL, 扬州大学 教师个人主页服务平台, A771726在制备治疗流感病毒感染相关疾病的药物中的应用徐秀龙, teacher.yzu.edu.cn/XXL1/en/zlc...
实用临床医药杂志
肌酸激酶评估 糖尿病 酮症酸中毒患者临床结局的价值 赵永玲, 郑超, 杨晓荣 2020, 24(21): 73-75,85. doi: 10.7619/jcmp.202021021 摘要(201) PDF (654KB)(11) 摘要: 目的 ... 实用临床医药杂志
ref=" http://www. baidu.com/link? url=4ATSXIDxjyg0ulkGBuuILmSGSrYOnbUTpSCYbB3S-KjWYCXf2cQ5okl_EJW8KSieiHtNbzldEyCNe1LrsvkV64e9xh2-FGQLufEqvZdMrsa ">扬州大学教师个人主页服务平台 XXL--Home--Scientific Res...
XXL , A771726在制备治疗流感病毒感染相关疾病的药物中的应用 XXL , 来氟米特 或其活性代谢产物A771726在制备治疗2型 糖尿病 的药物中的应用 Published Books No content Resea... teacher.yzu.edu.cn/XXL1/en/zhy...
实用临床医药杂志
目的 分析 糖尿病 肾病患者不同阶段生长分化因子-15(GDF-15)表达的变化及临床意义.方法 抽取150例 糖尿病 肾病患者,包括50例白蛋白尿正常患者(A组)、50例少量白蛋白尿患者(B组)和... 实用临床医药杂志
ref=" http://www. baidu.com/link? url=te0Y02T7IHNSWHZ3qSy6_OvT8gxpD2f5IeM7N0D0QQzEpLUshx9509yEchDU9c2K-WLidDh_Bv8EbBh7YsAoRx9__FwMWoesREqI_iktV8K ">扬州大学教师个人主页服务平台 徐秀龙--中文主页--专利
徐秀龙 首页 科学研究 教学研究 获奖信息 招生信息 学生信息 我的相册 教师博客 微信扫描 当前位置:中文主页>科学研究>专利 专利 徐秀龙 , A771726在制备治疗流感病毒感染相... teacher.yzu.edu.cn/XXL1/zh_CN/...
ref=" http://www. baidu.com/link? url=4RppJVRMLUmdvZ_7UTfFgqdKa_D31lJSXDTMFky_mLfcxvpjK7bokD6Pk8ukEeYkfZP-3PVBO3EExoPnaKCLTq ">扬州大学教师个人主页服务平台 徐秀龙--中文主页--专利
扬州大学 官网手机版徐秀龙 首页 科学研究研究领域 论文成果 专利 著作成果 科研项目 教学研究 教学资源 授课信息 教学...徐秀龙 , 来氟米特 或其活性代谢产物A771726在制备治... teacher.yzu.edu.cn/XXL1/zh_CN/...
实用临床医药杂志
肌酸激酶评估 糖尿病 酮症酸中毒患者临床结局的价值 赵永玲, 郑超, 杨晓荣 2020, 24(21): 73-75,85. doi: 10.7619/jcmp.202021021 摘要(217) PDF (654KB)(11) 摘要: 目的 ... 实用临床医药杂志


参考
- ^ 基于S6K1靶点的抗2型糖尿病和抗肥胖的药物作用机理研究 陈俊红 扬州大学 知网 http://cdmd.cnki.com.cn/article/cdmd-11117-1018091490.htm
- ^ 陈俊红. 基于S6K1靶点的抗2型糖尿病和抗肥胖的药物作用机理研究[D].扬州大学,2018. https://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CDFD&dbname=CDFD&filename=1018091490.nh
- ^ 陈俊红.基于S6K1靶点的抗2型糖尿病和抗肥胖的药物作用机理研究.2018.扬州大学,PhD dissertation. https://d.wanfangdata.com.cn/thesis/Y3431767
- ^ 百度学术:陈俊红.(2018).基于S6K1靶点的抗2型糖尿病和抗肥胖的药物作用机理研究(博士学位论文,扬州大学). https://xueshu.baidu.com/usercenter/paper/show?paperid=145q0660vp3h0ax0bx5r0xj04k143408
- ^ 百度学术:陈俊红.基于S6K1靶点的抗2型糖尿病和抗肥胖的药物作用机理研究[D].导师:徐秀龙.扬州大学,2018. https://xueshu.baidu.com/usercenter/paper/show?paperid=eebfac294e7977593fe96198779edc5d
- ^ 百度学术:SrcDatabase-来源库: 博士 Title-题名: 基于S6K1靶点的抗2型糖尿病和抗肥胖的药物作用机理研究 Author-作者: 陈俊红 Organ-单位: 扬州大学 Source-文献来源: 扬州大学 https://xueshu.baidu.com/usercenter/paper/show?paperid=1w500xw08j350cs0au1r0a30cg457292
- ^ Chen, Junhong, Jing Sun, Michelle E Doscas, Jin Ye, Ashley J Williamson, Yanchun Li, Yi Li, Richard A Prinz, and Xiulong Xu. "Control of hyperglycemia in male mice by leflunomide: mechanisms of action". Journal of Endocrinology 237.1 (2018): 43-58. < https://doi.org/10.1530/JOE-17-0536>. Web. 3 Aug. 2022. https://joe.bioscientifica.com/view/journals/joe/237/1/JOE-17-0536.xml
- ^ Chen, Junhong, Jing Sun, Michelle E Doscas, Jin Ye, Ashley J Williamson, Yanchun Li, Yi Li, Richard A Prinz, and Xiulong Xu. "Control of hyperglycemia in male mice by leflunomide: mechanisms of action", Journal of Endocrinology 237, 1 (2018): 43-58, accessed Aug 3, 2022, https://doi.org/10.1530/JOE-17-0536
- ^ PDF下载:Chen, J., Sun, J., Doscas, M. E., Ye, J., Williamson, A. J., Li, Y., Li, Y., Prinz, R. A., & Xu, X. (2018). Control of hyperglycemia in male mice by leflunomide: mechanisms of action, Journal of Endocrinology, 237(1), 43-58. Retrieved Aug 3, 2022, from https://joe.bioscientifica.com/view/journals/joe/237/1/JOE-17-0536.xml https://joe.bioscientifica.com/downloadpdf/journals/joe/237/1/JOE-17-0536.pdf
- ^ 百度学术:Zhang, Jingting et al. “Role of c-Jun terminal kinase (JNK) activation in influenza A virus-induced autophagy and replication.” Virology vol. 526 (2019): 1-12. doi:10.1016/j.virol.2018.09.020 https://xueshu.baidu.com/usercenter/paper/show?paperid=1d6y0tg07q2w0mu057600jh0ws235295
- ^ 谷歌镜像 https://google.icloudnative.io/
- ^ 扬州大学教师个人主页服务平台 徐秀龙--中文主页--专利 http://teacher.yzu.edu.cn/XXL1/zh_CN/zlcg/337479/list/index.htm
- ^ a b @罕见病友临行疾呼 注:A771726 即特立氟胺 博主所服用药物
- ^ 榆林市卫生健康委员会 类风湿消炎药也可降糖 索引号:013299 主题分类:疾病防控来源:生命时报 发布时间:2018-03-23 16:09:44 http://wjw.yl.gov.cn/index.php?m=Article&a=show&id=13299
- ^ 榆林市卫生健康委员会 类风湿消炎药也可降糖 索引号:013294主题分类:疾病防控来源:生命时报 发布时间:2018-03-23 16:05:06 http://wjw.yl.gov.cn/index.php?m=Article&a=show&id=13294
- ^ @罕见病友临行疾呼 注:爱若华®长征新开 等是通过一致性评价的来氟米特
- ^ 厉害了!我校徐秀龙教授课题组糖尿病研究实现新突破 扬州大学校友会 微信公众号2018-03-19 18:11 发表于江苏 https://mp.weixin.qq.com/s/Bg35adly3DqP1EHNaDbEHw
- ^ Zhang J, Ruan T, Sheng T, Wang J, Sun J, Wang J, Prinz RA, Peng D, Liu X, Xu X. Role of c-Jun terminal kinase (JNK) activation in influenza A virus-induced autophagy and replication. Virology. 2019 Jan 2;526:1-12. doi: 10.1016/j.virol.2018.09.020. Epub 2018 Oct 10. PMID: 30316042; PMCID: PMC6424123. (Xiulong Xu) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6424123/
- ^ 提问者链接不可访问 https://k.sina.cn/article_1659674142_62ec9e1e020005jcv.html
- ^ @罕见病友临行疾呼 回答:如何看待扬州大学关于来氟米特在糖尿病治疗上的研究? https://www.zhihu.com/question/269153929/answer/2607493605
- ^ 百度搜索 site:sina.cn 扬州大学 来氟米特 糖尿病 https://www.baidu.com/s?wd=site%3Asina.cn%20扬州大学%20来氟米特%20糖尿病
- ^ 5种常见的疾病根本无法治愈,说能治愈的或只想骗钱,别上当受骗 2021年07月27日 20:51 新浪网 作者 奇妙的本草 http://k.sina.com.cn/article_7021488421_1a2836925001011s01.html
- ^ 百度搜索 "扬州大学" "来氟米特" "糖尿病" https://www.baidu.com/s?wd="扬州大学"%20"来氟米特"%20"糖尿病"
- ^ 百度搜索 "扬州大学" "来氟米特" "糖尿病" site:gov.cn https://www.baidu.com/s?wd="扬州大学"%20"来氟米特"%20"糖尿病"%20site%3Agov.cn
- ^ 百度搜索 "扬州大学" "来氟米特" "糖尿病" site:edu.cn https://www.baidu.com/s?wd="扬州大学"%20"来氟米特"%20"糖尿病"%20site%3Aedu.cn
编辑于 2022-08-03 17:57
・IP 属地四川