

TCGAbiolinks使用
10 个月前

1. 数据下载
####1.1 加载包
rm(list=ls()) #清除前面工作内容
library(TCGAbiolinks) #数据下载与准备
library(tidyverse)
library(limma)
library(SummarizedExperiment)
library(EDASeq) #EDASeq软件包标准化mRNA转录本和miRNA,
library(edgeR) #用于差异分析
####(1.2)GDCquery
####GDCquery()筛选需要的数据,TCGAbiolinks包下载TCGA数据进行表达差异分析
query <- GDCquery(project = "TARGET-OS",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts")
GDCdownload(query)
samplesrda <- GDCprepare(query = query)
2.1 提取counts和tpm
countsdata<- assay(samplesrda,i = "unstranded") #tpm_unstrand fpkm_unstrand
tpmdata<- assay(samplesrda,i = "tpm_unstrand")
geneid22=data.frame(id=rowData(samplesrda)@listData[["gene_id"]], gene_name= rowData(samplesrda)@listData[["gene_name"]],gene_type=rowData(samplesrda)@listData[["gene_type"]])
2.2 去除
counts=cbind(geneid22,countsdata)
tpm=cbind(geneid22,tpmdata)
a1=gsub("_PAR_Y","",counts$id)
a2=gsub("_PAR_Y","",tpm$id)
counts2=transform(counts, id=a1)
colnames(counts2)=colnames(counts)
tpm2=transform(tpm, id=a2)
colnames(tpm2)=colnames(tpm)
counts3=distinct(counts2,id,.keep_all = T)
tpm3=distinct(tpm2,id,.keep_all = T)
2.3 取基因名平均值,基因名改为行名
counts4=counts3[,-c(1,3)]
counts5= avereps(counts4[,-1],ID = counts4$gene_name)
tpm4=tpm3[,-c(1,3)]
tpm5= avereps(tpm4[,-1],ID = tpm4$gene_name)
2.4 去除低丰度基因
countsdata=counts5[which(apply(counts5,1,function(x){return(sum(x>10))})>ncol(counts5)*0.25),] #25%样本表达>10的基因
tpmdata=tpm5[row.names(tpm5)%in%countsdata]
save(countsdata,file = 'countsdata.Rdata')
save(tpmdata,file = 'tpmdata.Rdata')
####(1.3)查询分类
####getResults(query, rows, cols)根据指定行名或列名从query中获取结果,此处用来获得样本的barcode
#### 此处共检索出407个barcodes
samplesDown <- getResults(query,cols=c("cases"))
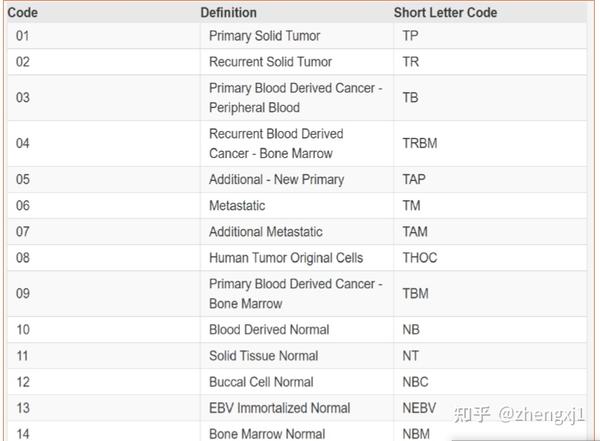
# 从samplesDown中筛选出TP(实体肿瘤)样本的barcodes
# TCGAquery_SampleTypes(barcode, typesample)
# TP代表PRIMARY SOLID TUMOR;
# NT-代表Solid Tissue Normal(其他组织样本可参考学习文档)
##此处共检索出375个TP样本barcodes
samplesTP <- TCGAquery_SampleTypes(barcode = samplesDown,
typesample = "TP")
####从samplesDown中筛选出NT(正常组织)样本的barcode 32个NT样本barcodes
samplesNT <- TCGAquery_SampleTypes(barcode = samplesDown,
typesample = "NT")
####(1.4) 设置barcodes参数,筛选符合要求的375个肿瘤样本数据和32正常组织数据
queryDown <- GDCquery(project = "TCGA-STAD",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts",
barcode = c(dataSmTP, dataSmNT))
####1.1 下载数据,默认存放位置为当前工作目录下的GDCdata文件夹中。
GDCdownload(queryDown,
method = "api",
directory = "GDCdata",
files.per.chunk = 6)
#method ;"API"或者"client"。"API"速度更快,但是容易下载中断。
#directory:下载文件的保存地址。Default: GDCdata。
#files.per.chunk = NULL:使用API下载大文件的时候,可以把文件分成几个小文件来下载,可以解决下载容易中断的问题。
gdcdata=function(i){
library(TCGAbiolinks)
projects <- getGDCprojects()
library(dplyr)
projects <- projects %>%
as.data.frame() %>%
select(project_id,tumor) %>%
filter(grepl(pattern="TCGA",project_id))
## 0.运行信息
print(paste0("Downloading number ",i,",project name: ",projects$project_id[i]))
## 1.查询信息
query.exp = GDCquery(project = projects$project_id[i],
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts")
## 2.正式下载
GDCdownload(query.exp)
## 3.多个数据合并
pre.exp = GDCprepare(query = query.exp)
## 4.提取表达量数据
library(SummarizedExperiment)
countsdata = SummarizedExperiment::assay(pre.exp,1)
fpkmdata=SummarizedExperiment::assay(pre.exp,5)
tpmdata=SummarizedExperiment::assay(pre.exp,4)
gene_id=data.frame(id=rowData(pre.exp)@listData[["gene_id"]], gene_name= rowData(pre.exp)@listData[["gene_name"]],gene_type=rowData(pre.exp)@listData[["gene_type"]])
counts=cbind(gene_id,countsdata)
fpkm=cbind(gene_id,fpkmdata)
tpm=cbind(gene_id,tpmdata)
#临床信息
clinical <- GDCquery_clinic(project = projects$project_id[i], type = "clinical")
## 5.保存数据
filename1 = paste0("result/",projects$project_id[i],"-counts.txt")
filename2 = paste0("result/",projects$project_id[i],"-fpkm.txt")
filename3 = paste0("result/",projects$project_id[i],"-tpm.txt")
filename4 = paste0("result/",projects$project_id[i],"-clinical.txt")
write.table(counts,filename1,sep="\t",col.names=T,row.names=F,quote=F)
write.table(fpkm,filename2,sep="\t",col.names=T,row.names=F,quote=F)
write.table(tpm,filename3,sep="\t",col.names=T,row.names=F,quote=F)
write.table(clinical,filename4,sep="\t",col.names=T,row.names=F,quote=F)
dir.create("result")
for (i in 1:33) {
gdcdata(i)
________________________________________________________________________________________________________________
或者可以先不将queryDown分组,后面再分组,如下
#数据查询与下载
library(TCGAbiolinks)
query <- GDCquery(project = "TCGA-COAD",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - FPKM-UQ")
GDCdownload(query)
#数据准备
samplespre <- GDCprepare(query, save = TRUE,
save.filename = "samplespre_summerizedExperiment.rda",remove.files.prepared = TRUE)
#数据标准化与筛选
library(SummarizedExperiment)
samples.information=colData(samplespre)
sampleNorm <- TCGAanalyze_Normalization(tabDF = samplespre, geneInfo = geneInfoHT)
sampleFilt <- TCGAanalyze_Filtering(tabDF = sampleNorm,method ="quantile",qnt.cut = 0.25)
#f数据分组
samplesNT <- TCGAquery_SampleTypes(barcode = colnames(sampleFilt),typesample = c("NT"))
samplesTP <- TCGAquery_SampleTypes(barcode = colnames(sampleFilt),typesample = c("TP"))
#差异分析
dataDEGs <- TCGAanalyze_DEA(mat1 =sampleFilt[,samplesNT],
mat2 = sampleFilt[,samplesTP],
Cond1type = "Normal",
Cond2type = "Tumor",
fdr.cut = 0.05,
logFC.cut = 1,
method = "glmLRT")
#整理导出
dataDEGsFiltLevel <- TCGAanalyze_LevelTab(dataDEGs,"Tumor","Normal",
dataFilt[,samplesTP],dataFilt[,samplesNT])
write.csv(dataDEGsFiltLevel,file="DEGs.csv")


2. 数据准备
####3.1:GDCprepare()将前面GDCquery()的结果准备成R语言可处理的SE(SummarizedExperiment)文件
####读取下载的数据并将其准备到R对象中,在工作目录生成(save=TRUE)STAD_case.rda文件
#### GDCprepare():Prepare GDC data,准备GDC数据,使其可用于R语言中进行分析
samplesrda <- GDCprepare(query = queryDown, save = TRUE, save.filename =
"samplesrda.rda")

3. TCGAanalyze_Preprocessing()对数据进行预处理
####使用spearman相关系数去除数据中的异常值
# 去除dataPrep1中的异常值,dataPrep1数据中含有肿瘤组织和正常组织的数据
# TCGAanalyze_Preprocessing(object, cor.cut = 0, filename = NULL,
# width = 1000, height = 1000, datatype = names(assays(object))[1])
# 函数功能描述:Array Array Intensity correlation (AAIC) and correlation boxplot to define outlier
samplespre <- TCGAanalyze_Preprocessing(object = samplesrda,
cor.cut = 0.6,
datatype = "HTSeq - Counts")
#将预处理后的数据samplespre,写入新文件“samplespre.csv”
write.csv(samplespre,file = "samplespre.csv",quote = FALSE)4. TCGAtumor_purity()筛选肿瘤纯度大于60%的肿瘤barcodes
# TCGAtumor_purity(barcodes, estimate, absolute, lump, ihc, cpe),
# 使用来自5种方法的5个估计值作为阈值对TCGA样本进行过滤,
# 这5个值是estimate, absolute, lump, ihc, cpe,
# 这里设置cpe=0.6(cpe是派生的共识度量,是将所有方法的标准含量归一化后的均值纯度水平,以使它们具有相等的均值和标准差)
# 筛选肿瘤纯度大于等于60%的样本数据
samplespurity <- TCGAtumor_purity(colnames(samplespre), 0, 0, 0, 0, 0.6)
####这里筛选的60%的样本数据并没有用成功(没有肿瘤纯度的信息),然后直接赋值
# filtered 为被过滤的数据, pure_barcodes是我们要的肿瘤数据
####过滤的数据太多,重新分配
samplespurity$pure_barcodes <- samplesTP
samplespurity$filtered <- samplesNT
tumorsamples <- samplespurity$pure_barcodes
normalsamples <- samplespurity$filtered5. 将肿瘤表达矩阵与正常组织表达矩阵合并,进行基因注释
#5.1 表达矩阵合并
###肿瘤表达矩阵与正常组织表达矩阵合并
combinedsamples <-samplespre[,c(tumorsamples,normalsamples)]

#5.2 基因注释, 需要加载“SummarizedExperiment”包,
#“SummarizedExperiment container”每个由数字或其他模式的类似矩阵的对象表示。
# 行通常表示感兴趣的基因组范围和列代表样品。
rownames(combinedsamples)<-rowData(samplesrda)$external_gene_name
write.csv(combinedsamples,file = "combinedsamples.csv",quote = FALSE)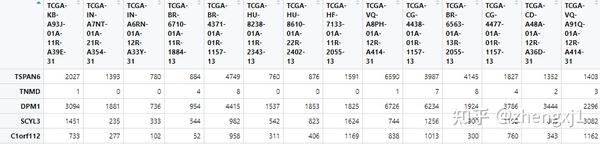

6. 表达矩阵标准化和过滤
####6.1 TCGAanalyze_Normalization()`使用EDASeq软件包标准化mRNA转录本和miRNA。
samplesNorm <- TCGAanalyze_Normalization(tabDF = combinedsamples,
geneInfo = geneInfo,
method = "gcContent")
####6.2 将标准化后的数据再过滤,去除掉表达量较低(count较低)的基因,得到最终的数据
samplesFilt <- TCGAanalyze_Filtering(tabDF = samplesNorm,
method = "quantile",
qnt.cut = 0.25)
7. 差异分析
##定义样本的分组,前375个是肿瘤样本,后32个是正常样本
#7.1 定义肿瘤样本分组并取log
mat1 <- dataFilt_STAD_final[,dataSmTP]
mat1 <- log(mat1+1)
#7.2 定义正常组织样本分组并取log
mat2 <- dataFilt_STAD_final[,dataSmNT]
mat2 <- log(mat2+1)
#7.3 差异分析
Data_DEGs <- TCGAanalyze_DEA(mat1 = mat1,
mat2 = mat2,
Cond1type = "Tumor",
Cond2type = "Normal",
pipeline="limma",
batch.factors = c("TSS"),
voom = TRUE,
contrast.formula = "Mycontrast=Tumor-Normal")
# 7.4 如何导出???
write.csv(mat1,file = "mat1.csv",quote = FALSE)
write.csv(mat2,file = "mat2.csv",quote = FALSE)8. 富集分析
#8.1 设置logFC,挑选表达有差异的基因进行富集分析
Data_DEGs_high_expr <- Data_DEGs[Data_DEGs$logFC >=1,]
Genelist <- rownames(Data_DEGs_high_expr)
ansEA <- TCGAanalyze_EAcomplete(TFname="DEA genes Normal Vs Tumor",
Genelist)
#8.2 富集分析可视化
TCGAvisualize_EAbarplot(tf = rownames(ansEA$ResBP),
GOBPTab = ansEA$ResBP,
GOCCTab = ansEA$ResCC,
GOMFTab = ansEA$ResMF,
PathTab = ansEA$ResPat,
nRGTab = Genelist,
nBar = 10, #显示条形图的数量
filename = "TCGAvisualize_EAbarplot_Output.pdf")
1. 下载临床数据
clinical <- GDCquery_clinic("TCGA-STAD", "clinical",save.csv = FALSE )2. 生存分析
###TCGAbiolinks包中自带的进行生存分析的函数是TCGAanalyze_survival,但是我们也有其他的包可以进行生存分析,在第二部分中会进行对比说明
####TCGAanalyze_survival 的用法:
####利用得到的临床数据探索性别对患者生存的影响
TCGAanalyze_survival(clin.STAD,
clusterCol="gender",
risk.table = F,
xlim = c(100,1000),
ylim = c(0.4,1),
conf.int = T,
color = c("Dark2"))3. 探索基因表达对生存的影响
####随机选取PGA5基因为例探索单个基因表达的情况对患者生存的影响
# 1、取特定基因(这里以PGA5基因为例)在癌症样本中的表达
# 2、先读取基因表达矩阵
# 3、提取特定基因在癌症样本中的表达,选出的样本都是肿瘤样本
samplesTP <- TCGAquery_SampleTypes(colnames(dataFilt_STAD_final), typesample = c("TP"))
PGA5 <- dataFilt_STAD_final[c("PGA5"),samplesTP]
View(PGA5)
# 4、修改样本名称(原是TCGA-DD-AAD5-01A-11R-A41C-07),
# 但是clin.STAD数据中的样本名称是12位,
# 因为后期要把 clin.STAD 数据和表达数据结合起来,需要将名字简化成TCGA-DD-AAD5
names(PGA5) <- sapply(strsplit(names(PGA5),'-'),function(x) paste0(x[1:3],collapse="-"))
PGA5 <-as.data.frame(cbind(names(PGA5),PGA5))
colnames(PGA5) <- c("submitter_id","PGA5")
# 5、合并PGA5基因与临床数据
clin.STAD <- merge(clin.STAD,PGA5,by="submitter_id")
View(clin.STAD)
# 6、从 clin.STAD 中选取进行生存分析需要的数据组成新的
##数据框(有barcode、生存状态、死亡时间、随访时间、ABCB1基因的表达数据)
df<-subset(clin.STAD,select =c(submitter_id,vital_status,days_to_death,days_to_last_follow_up,PGA5))
View(df)
# 7、去除PGA5基因表达数据缺失的样本
#去掉 NA
df <- df[!is.na(df$PGA5),]
# 8、根据ABCB1基因的表达情况进行分组,取基因的平均值,大于平均值的为H,小于为L
df$PGA5 <- as.numeric(df$PGA5)
df$exp <- ''
df[df$PGA5 >= mean(df$PGA5),]$exp <- "H"
df[df$PGA5 < mean(df$PGA5),]$exp <- "L"
# 9、用 TCGAanalyze_survival 函数进行生存分析
TCGAanalyze_survival(df,
clusterCol="exp",
risk.table = FALSE,
conf.int = FALSE,
color = c("Dark2"))
# 10、除了使用TCGAanalyze_survival 函数,我们还可以使用 survival、survminer这两个包来进行生存分析。我们可以来对比一下他们的区别。
# 10.1 用status表示患者结局,1表示删失,2表示死亡
df2 <- df
# 10.2 将status表示患者结局,1表示删失,2表示死亡
df2[df2$vital_status=='Dead',]$vital_status <- 2
df2[df2$vital_status=='Alive',]$vital_status <- 1
df2$vital_status <- as.numeric(df2$vital_status)
df2$time <- df2$days_to_death
df2$time[which(is.na(df2$time))] <- df2$days_to_last_follow_up[which(is.na(df2$time))]
View(df2)
library(survival)
library(survminer)
fit <- survfit(Surv(time, vital_status)~exp, data=df2) # 根据表达建模
# 显示P value
surv_pvalue(fit)$pval.txt
ggsurvplot(fit,pval=TRUE)https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files
library(dplyr)
library(stringr)
library(stringr)
> d1 <- read.table('test.txt', sep = '\t', header = TRUE)
tag t c g a
1 ENSG00000000003.13 2969 4725 1350 1667
2 ENSG00000000005.5 5 14 2 0
3 ENSG00000000419.11 1608 1588 749 888
安装biomaRt(因为我的R版本是3.5以上的。因此安装方式如下)
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.10")
安装biomaRt包
BiocManager::install(c("biomaRt"))
除ensemble_ID的版本号。version其实就是小数点后面的部分,ensembl_ID与其他基因ID进行转换时是不带有小数点的。
如果你拿到的数据有小数点,那就没法顺利的merge了,所以就要把它去掉。现在有两种方法可以直接使用。
> d1$ensemble_id=unlist(str_split(d1$tag,"[.]",simplify=T))[,1]
tag t c g a ensemble_id
1 ENSG00000000003.13 2969 4725 1350 1667 ENSG00000000003
2 ENSG00000000005.5 5 14 2 0 ENSG00000000005
3 ENSG00000000419.11 1608 1588 749 888 ENSG00000000419
对基因进行注释-获取gene_symbol,用bioMart对ensembl_id转换成gene_symbol
library(biomaRt)
1.显示一下能连接的数据库
listMarts()
biomart version
1 ENSEMBL_MART_ENSEMBL Ensembl Genes 99
2 ENSEMBL_MART_MOUSE Mouse strains 99
3 ENSEMBL_MART_SNP Ensembl Variation 99
4 ENSEMBL_MART_FUNCGEN Ensembl Regulation 99
这里我们选择ensembl数据库
2.用useMart函数选定数据库
plant<-useMart("ensembl")
3.用listDatasets()函数显示当前数据库所含的基因组注释
dataset
1 acalliptera_gene_ensembl
2 acarolinensis_gene_ensembl
3 acchrysaetos_gene_ensembl
4 acitrinellus_gene_ensembl
5 amelanoleuca_gene_ensembl
6 amexicanus_gene_ensembl
7 ampachon_gene_ensembl
8 anancymaae_gene_ensembl
9 applatyrhynchos_gene_ensembl
10 atestudineus_gene_ensembl
11 bbbison_gene_ensembl
12 bgrunniens_gene_ensembl
13 bihybrid_gene_ensembl
14 bmutus_gene_ensembl
15 bsplendens_gene_ensembl
16 btaurus_gene_ensembl
17 bthybrid_gene_ensembl
18 cabingdonii_gene_ensembl
19 capalliatus_gene_ensembl
20 caperea_gene_ensembl
21 catys_gene_ensembl
... 有很多
这里我们要获取的基因注释的基因是人类基因,所以选择hsapiens_gene_ensembl
4.用useDataseq()函数选定数据库中的基因组
>mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
##这条语句的意思是:选定ensembl数据库中的hsapiens_gene_ensembl基因组
5.选定我们需要获得的注释类型
用lsitFilters()函数查看可选择的类型,选定要获取的注释类型,以及已知注释的类型
listFilters(mart)
1 chromosome_name
2 start
3 end
4 band_start
5 band_end
6 marker_start
7 marker_end
8 encode_region
9 strand
10 chromosomal_region
11 with_ccds
12 with_chembl
13 with_clone_based_ensembl_gene
14 with_clone_based_ensembl_transcript
15 with_dbass3
16 with_dbass5
17 with_ens_hs_transcript
18 with_ens_hs_translation
19 with_entrezgene_trans_name
20 with_embl
21 with_arrayexpress
22 with_genedb
... 有很多
选择好数据库,基因组,要获得的注释类型,和已知的注释类型,就可以开始获取注释了
6.用getBM()函数获取注释
hg_symbols<- getBM(attributes=c('ensembl_gene_id','hgnc_symbol',"chromosome_name", "start_position","end_position", "band"), filters= 'ensembl_gene_id', values = d1$ensembl_gene_id, mart = mart)
结果如下:
ensembl_gene_id hgnc_symbol chromosome_name start_position end_position band
1 ENSG00000000003 TSPAN6 X 100627108 100639991 q22.1
2 ENSG00000000005 TNMD X 100584936 100599885 q22.1